Анализ секвенирования транскриптома
Следите за обновлениями и дополнениями
Если Вы заметили опечатки, или ссылка испортилась, пожалуйста, напишите мне.
Чтобы ознакомиться с командами,
использовавшимися при выполнении практикума, перейдите по ссылке.
Задача: картировать чтения, полученные в результате секвенирования транскриптома (человек, версия сборки генома hg19)
Дано:
1. Чтения, картирующиеся на участок хромосомы человека (получены путем секвенирования РНК).
Файлы с одноконцевыми чтениями в формате fastq лежат на kodomo в директории /P/y14/term3/block4/SNP/rnaseq_reads.
Для выполнения практикума была взята та же хромосома (третья), что изучалась при выполнении заданий практикума 11.
Для каждой хромосомы было взято два файла fastq - это 2 биологические реплики.
2. Разметка человеческого генома по версии Gencode19 для сборки hg19. Разметка в формате .gtf лежит на kodomo в директории /P/y14/term3/block4/SNP/rnaseq_reads
(gff и gtf формат - это почти одно и то же)
Часть I: подготовка чтений
0. Создание рабочей директории.
Для выполнения практикума была создвна папка в рабочей директории /nfs/srv/databases/ngs/pavel-kravchenko/pr12, в которую были скопированы необходимые файлы.
1. Анализ качества чтений.
Была использована локальная версия программы FastQC и выполнен анализ качества ридов.
Команда: fastqc chr3.fastq
Программа выдаёт несколько массивов информации о интересующих нас ридах:
Basic Statistics - общая информация о числе ридов, методе секвенирования, GC составе и средней длине прочтения.
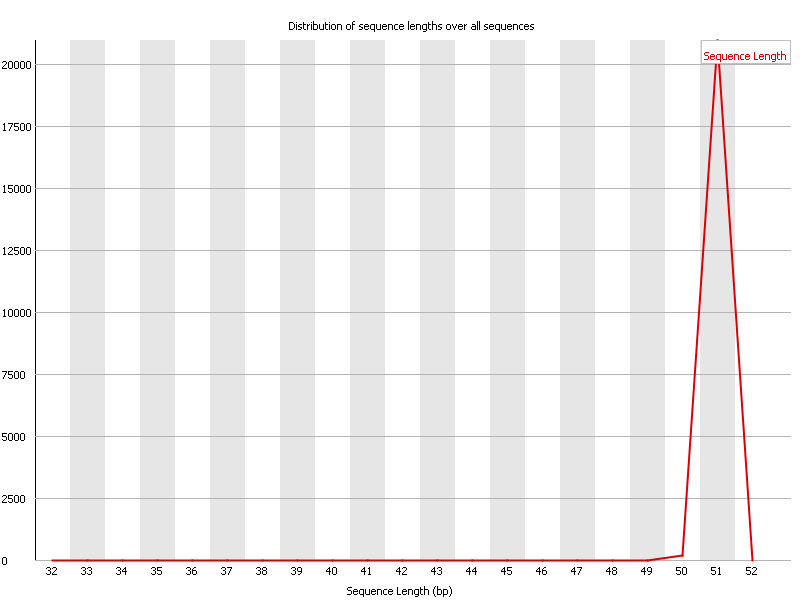
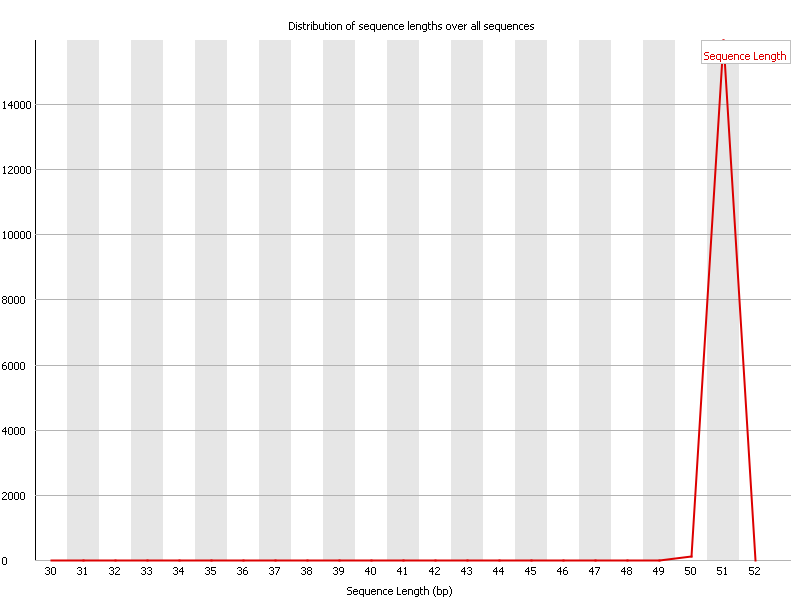
Так, для первой реплики было показано наличие 21211 ридов, секвенирование проводилось технологией Sanger / Illumina 1.9, длина ридов оказалась равной 33-51 нуклеотидам, а
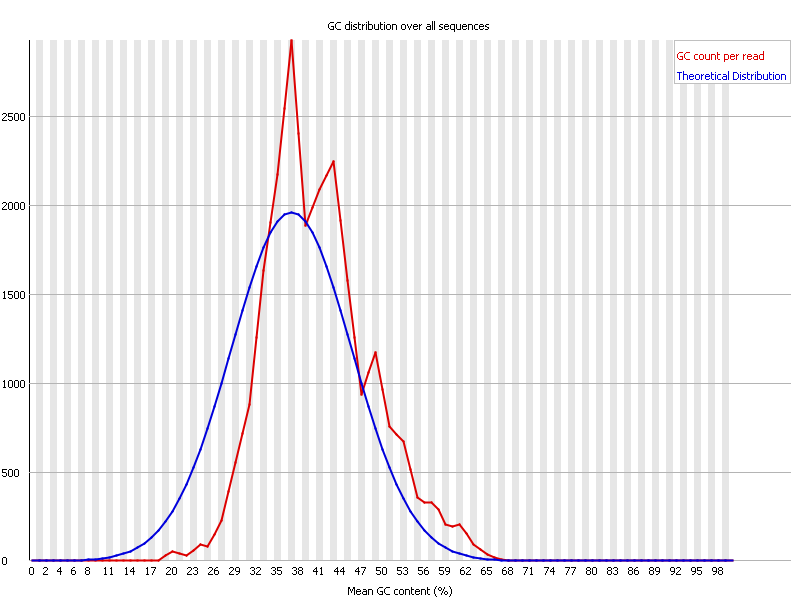
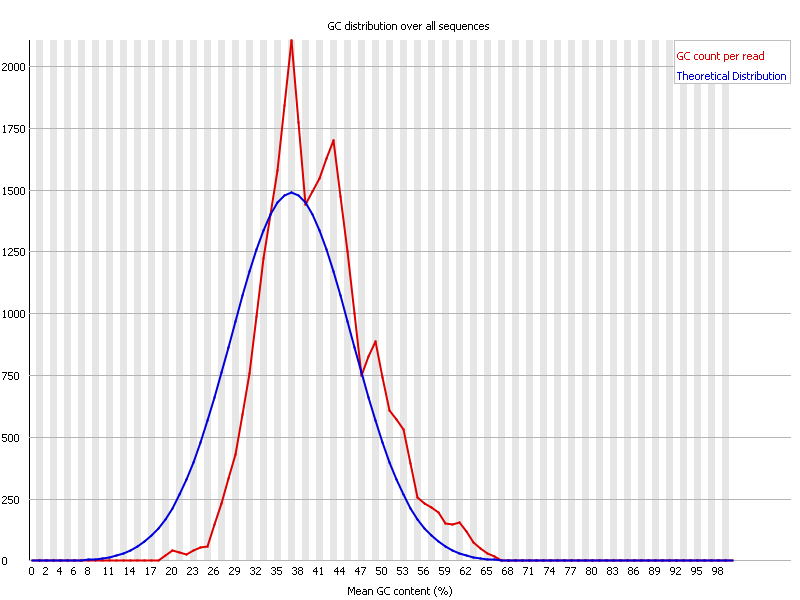
GC состав был определён значением в 41%.
Per base sequence quality - график качества ридов, который показывает, какого качества оказались те или иные прочтения.
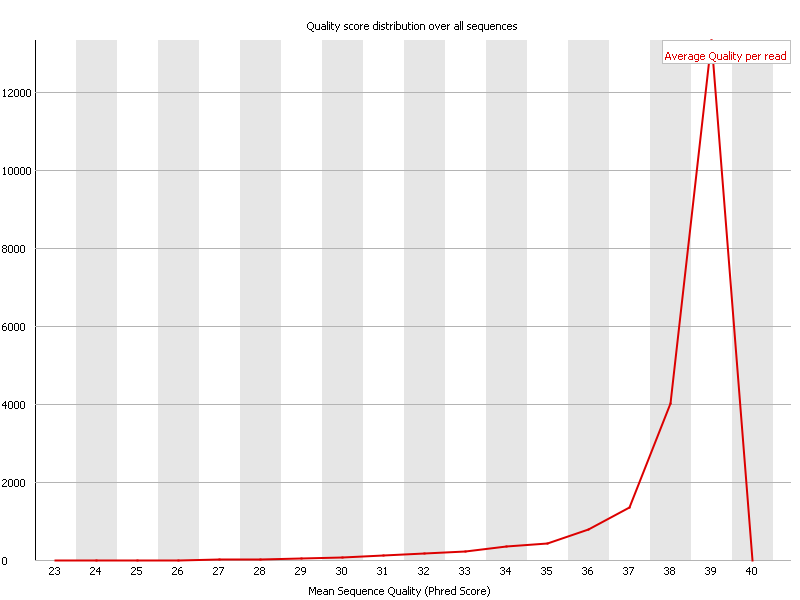
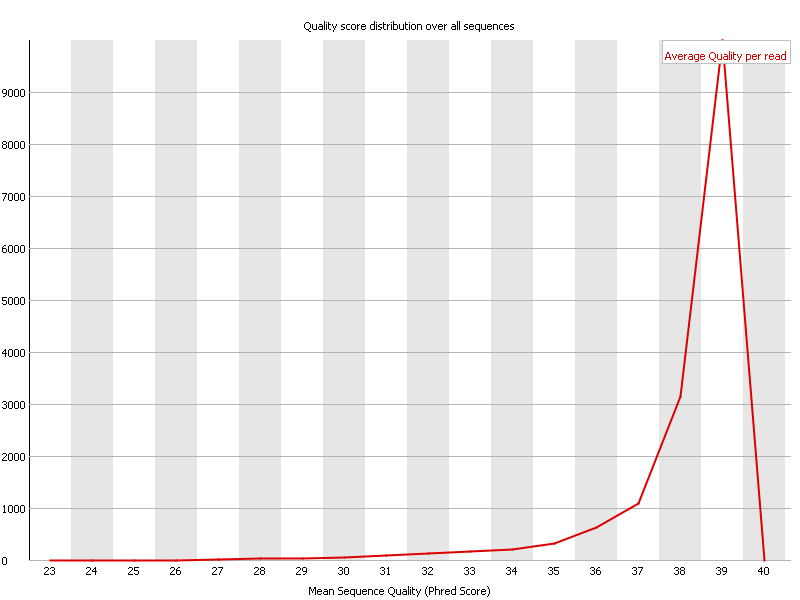
Per sequence quality scores - характеризует среднее качество прочтений. По графику можно понять, какое качество свойственно ридам, полученным после секвенирования.
Per base sequence content - данный график демонстрирует качество прочтения по нуклеотидам и может быть использован для оценки шума прибора и контроля качества прочтений. Также
можно посмотреть на частоту встречаемости того или иного нуклеотида в конкретной позиции ридов.
Per sequence GC content - показывает, какова средняя доля GC пар в ридах.
Sequence Length Distribution - среднее распределение длины ридов в файле.
Kmer Content - график, отражающий положение повторяющихся последовательностей в ридах.
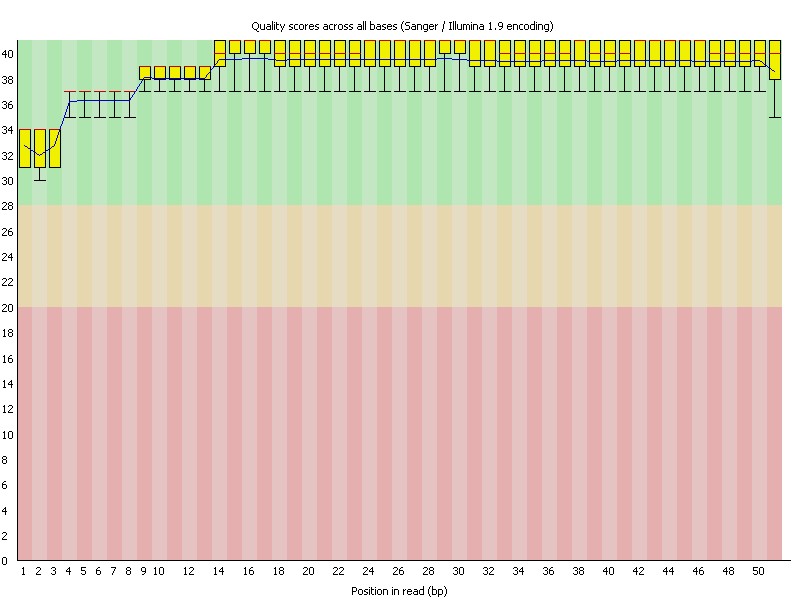
|
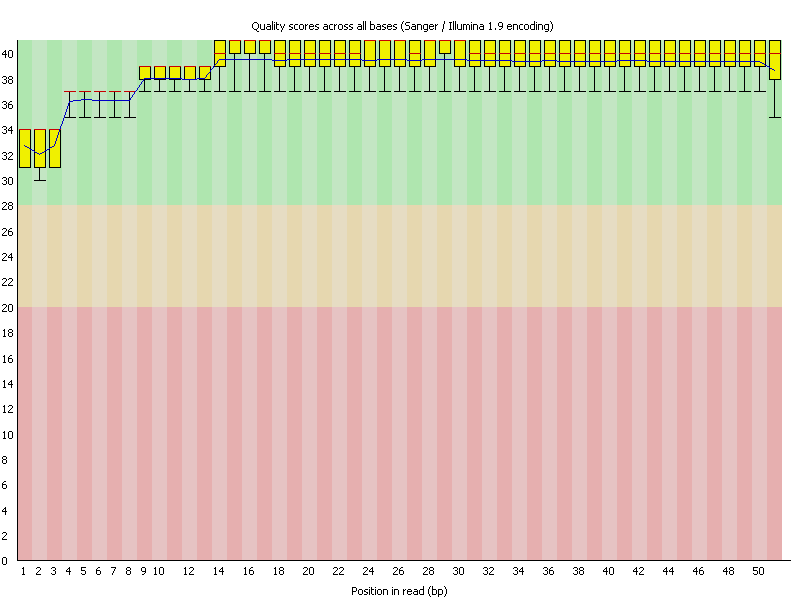
|
На рисунке 1 содержится информация о качестве ридов. Красным, в перделах бокса, отмечена медиана, синим - среднее качество прочтений. Жёлтые прямоугольники - это интерквартирльные размахи, то есть расстояние между нижней и верхней квартилью качества для каждой позиции прочтения. Зелёная зона имеет quality scores от 28 до 40, что является оптимальным для использования в генетическом анализе и интерпретировании результатов. Попадание в жёлтую и красную зоны говорит о более высоком уровне ошибок.
С помощью программы Trimmomatic, установленной на kodomo, была произведена очистка чтений с помощью программы Trimmomatic. Для этого с конца каждого чтения
были отрезаны нуклеотиды с качеством ниже 20 (TRAILING:20), и
оставлены только чтения длиной не меньше 50 нуклеотидов (MINLEN:50).
Команда: java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr3.fastq chr3_outfile.fastq TRAILING:20 MINLEN:35
Программа получила на вход 21211 ридов и, после очистки, выдала 21210 ридов, что составило ~100% от исходного их количества. Был отброшен 1 рид.
Так как все риды оказались короткими последовательностями приблизительно одного качества, не имеет смысла сравнивать очищенный и неочищенный файлы. Ниже будут приведены основные параметры содержащихся в файле первой реплики прочтений.
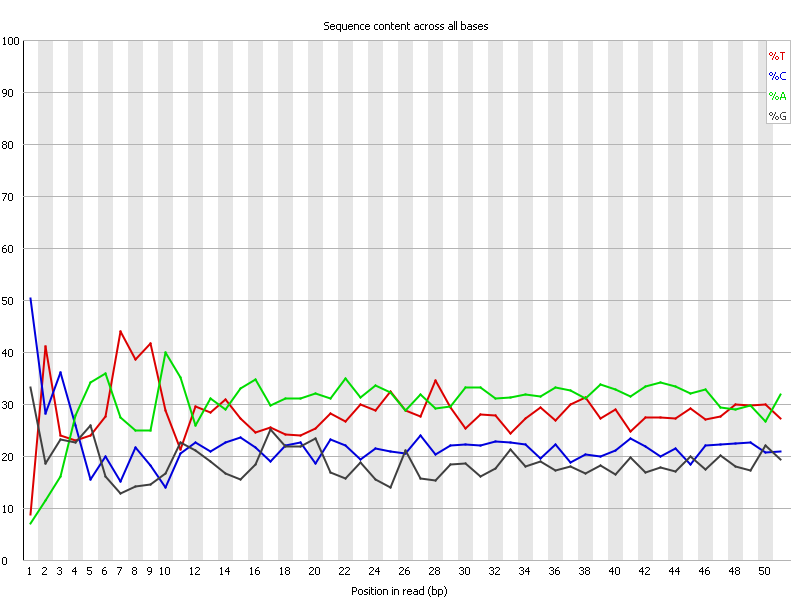
|
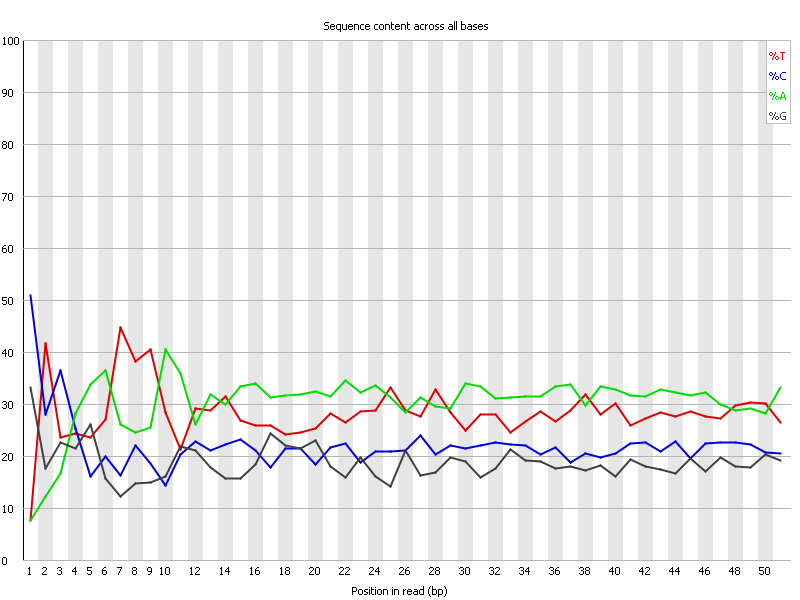
|
По зравнением с прошлым практикумом, заметно, что распределение нуклеотидов неравномерное.
|
|
|
|
|
|
|
|
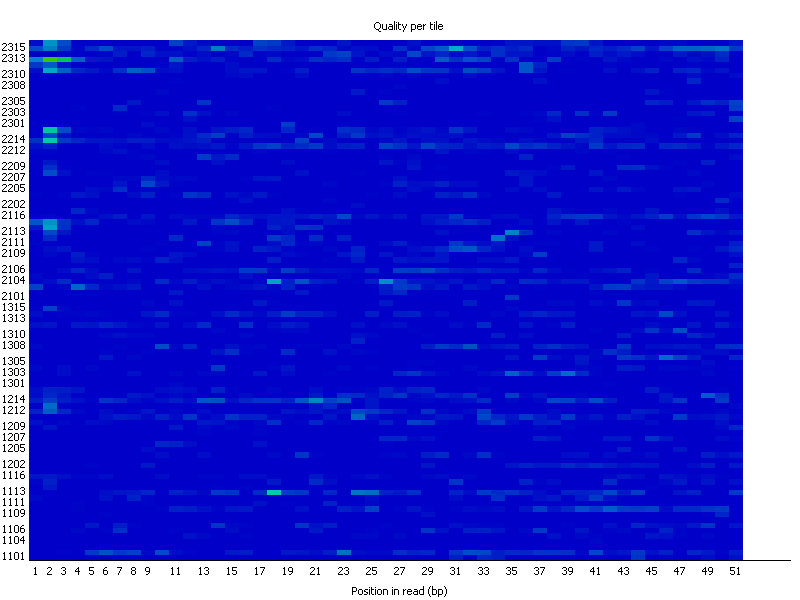
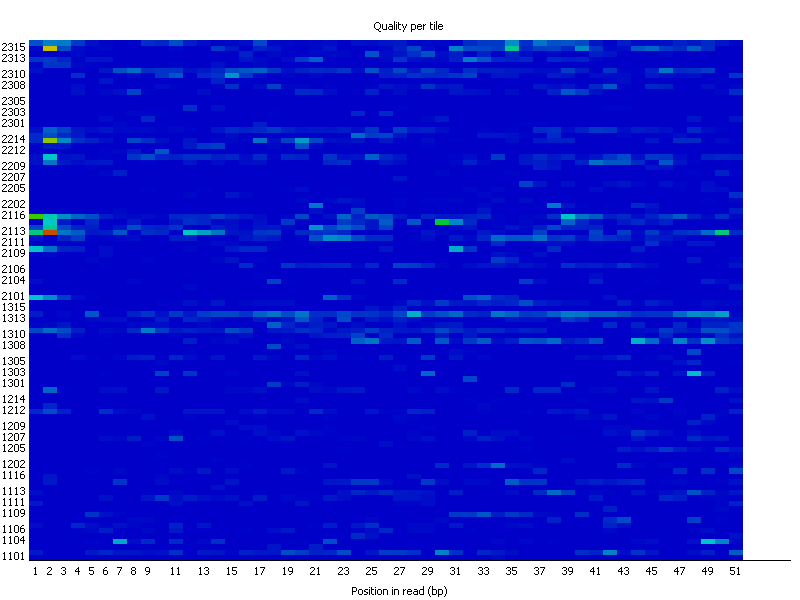
В то время как рисунки 2 - 5 заметно не отличаются друг от друга, тепловые карты на рисунке 6 демонстрируют различные по качеству проточные ячейки. Данная информация может быть успешно использована для отсечки шумов в столь дорогостоящем методе. Заметно, что во второй реплике шумов при секвенировании было больше (более тёплые области).
Часть II: картирование чтений
3. Картирование чтений.
Чтобы картировать прочтения, нужно произвести ряд манипуляций с командной строкой UNIX:
- Сначала необходимо проиндексировать референсную последовательность.
- Затем построить выравнивание прочтений и референса в формате .sam.
- Сохранить вывод программы hisat2 в отдельный файл.
Таблица 1. Использовавшиеся команды. Команда Результат export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 Прописывает временный путь до исполняемой программы hisat2-build chr3.fasta chr3 Индексирует референсную последовательность, записывает результат в *.ht2 файлы hisat2 -x chr3 -U chr3.[1/2].fastq --no-softclip > alignment[1/2].sam Строит выранивание прочтения и референса, записывает результат в .sam файл
Из конанды был убран параметр --no-spliced-alignment, так как при анализе транскриптома риды могут ложиться на два экзона разделённых интроном.
Программа hisat2 выдала следующую информацию о выравниваниях для первой реплики.
21210 reads; of these: 21210 (100.00%) were unpaired; of these: 156 (0.74%) aligned 0 times 20927 (98.67%) aligned exactly 1 time 127 (0.60%) aligned >1 times 99.26% overall alignment rate
Было поручено 21210 ридов, из которых 156 были выровнены 0 раз, а 20927 лишь один раз; 127 были выровнены более 1 раза.
И для второй реплики соответственно.
16133 reads; of these: 16133 (100.00%) were unpaired; of these: 139 (0.86%) aligned 0 times 15934 (98.77%) aligned exactly 1 time 60 (0.37%) aligned >1 times 99.14% overall alignment rate
Видно, что во второй реплике немного меньше ридов.
Был получен alignment1.sam файл с выравниванием ридов первой реплики и alignment2.sam файл с выравниванием ридов второй реплики
Таблица 2. Сводная таблица. Реплика Число ридов Число некартированных ридов Число картированых ровно 1 раз ридов Число картированых более 1 раза ридов Среднее качество выравнивания Первая 21210 156 20927 127 99.26% Вторая 16133 139 15934 60 99.14%
3. Анализ выравнивания.
Для анализа выравнивания использовалась программа samtools, работающая с бинарными файлами.
Таблица 3. Использовавшиеся команды. Команда Результат samtools view alignment[1/2].sam -b -o alignment[1/2].bam Переводит выравнивание в бинарный формат samtools sort alignment[1/2].bam -T out_sort[1/2].txt -o alignment_sorted[1/2].bam Индексирует референсную последовательность, записывает результат в *.ht2 файлы samtools index alignment_sorted[1/2].bam Индексирует отсортированный .bam файл samtools idxstats alignment_sorted[1/2].bam > out[1/2].txt Записывает откартировавшиеся прочтения
Для первой реплики был получен файл alignment_sorted1.bam, в котором содержатся отсортированные выравнивания, необходимые для дальнейшей работы.Файл out1.txt содержит информацию о картировании ридов:
chr3 198022430 21075 0 * 0 0 156
Данная информация говорит о том, что на хромосому было картировано 21075 ридов, тогда как 156 ридов не было картировано. Длина хромосомы составила 198022430 нуклеотидов.
Для второй реплики был получен аналогичный alignment_sorted2.bam. Его, как и второй файл out2.txt можно расшифровать аналогично. Интересно будет визуализировать и посмотреть выравнивания рядом.
4. Подсчет чтений. Работа с Bedtools
С помощью программы Bedtools были проанализирования покрытия генов полученными ридами. Использовались полученные выше сортированные файлы.
Для работы с программой необходимо подавать на вход .bed файлы. С этой целью была применена команда bamtobed, которая получает на вход .bam файл, а выдаёт .bed соответственно.
Файл с разметкой генов для следующих манипуляций по версии Gencode для генома человека сборки hg19 лежит по адресу: /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed
Программа bedtools может быть вызвана так: export PATH=${PATH}:/P/y14/term3/block4/SNP/bedtools2/bin
С помощью команды intersect, при использовании разметки генов для генома человека, было найдено пересечение полученного выравнивания с генами. Параметры запуска можно найти в подробном мануале.
Таблица 4. Использовавшиеся команды. Команда Результат bedtools bamtobed -i alignment_sorted[1/2].bam > chr3.[1/2]_s.bed Получает на вход .bam файл, а выдаёт .bed соответственно bedtools intersect -a gencode.genes.bed -b chr3.[1/2]_s.bed -u > chr3.[1/2]_crossed.bed Ищет перекрывание множества генов генома с множеством ридов выравнивания. -u отбираются лишь гены с ненулевым покрытием.
При обработке файлов было выдано предупреждение о отсутствии двух псевдогенов в множестве генов. Были получены файлы с перекрываниями обоих множеств.
Yes, My Lord:/nfs/srv/databases/ngs/pavel-kravchenko/pr12 bedtools intersect -a gencode.genes.bed -b chr3.1_s.bed -u > chr3.1_crossed.bed ***** WARNING: File gencode.genes.bed has inconsistent naming convention for record: GL877870.2 2348 2545 pseudogene CICP10 ***** WARNING: File gencode.genes.bed has inconsistent naming convention for record: GL877870.2 2348 2545 pseudogene CICP10
Визуализация в IGV
В программе IGV были визуализированы полученные файлы.
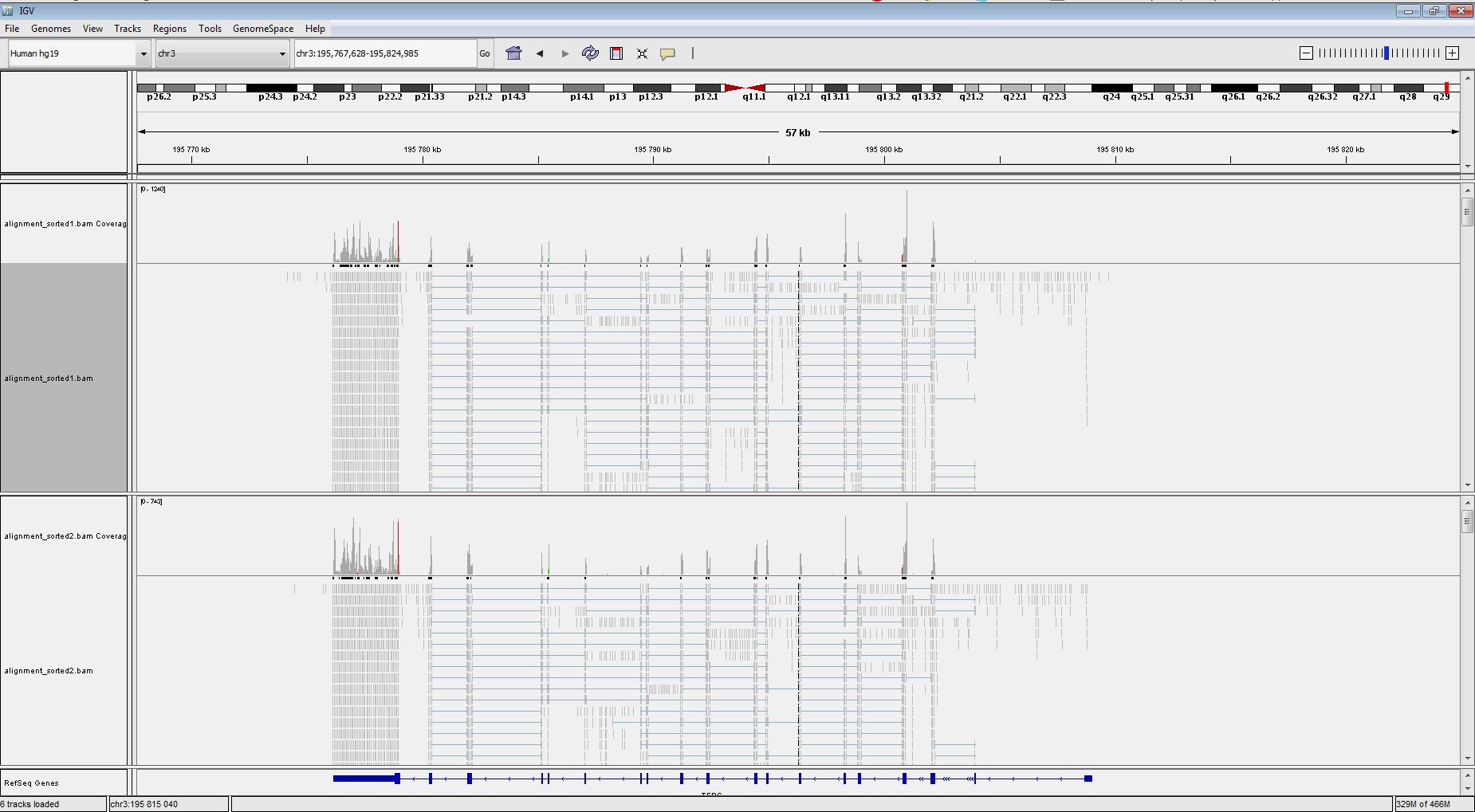
|
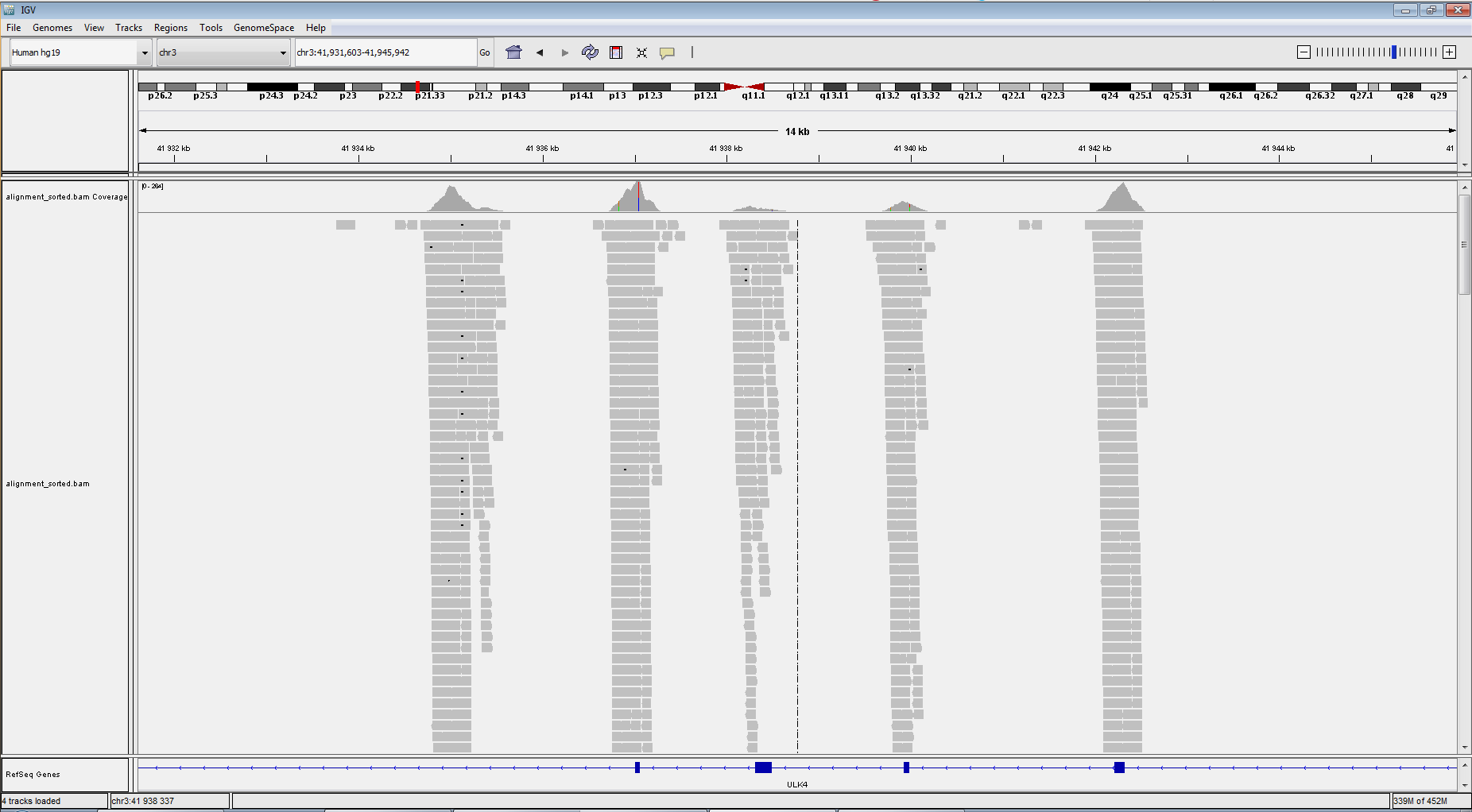
|
К сожалению, не удалось найти идентичный для обеих картинок участок, но в IGV видно, что при секвенировании SNP получают риды гораздо большей длины. Синими соединяющими линиями показаны участки, вырезающиеся во время сплайсинга. Можго заметить, что риды при экзомном секвенировании распологаются, в основном, упорядоченно, ровно над кодирующими областями. При поиске SNP используются праймеры, чтения с которых ложатся неравномерно.
5. Анализ результатов
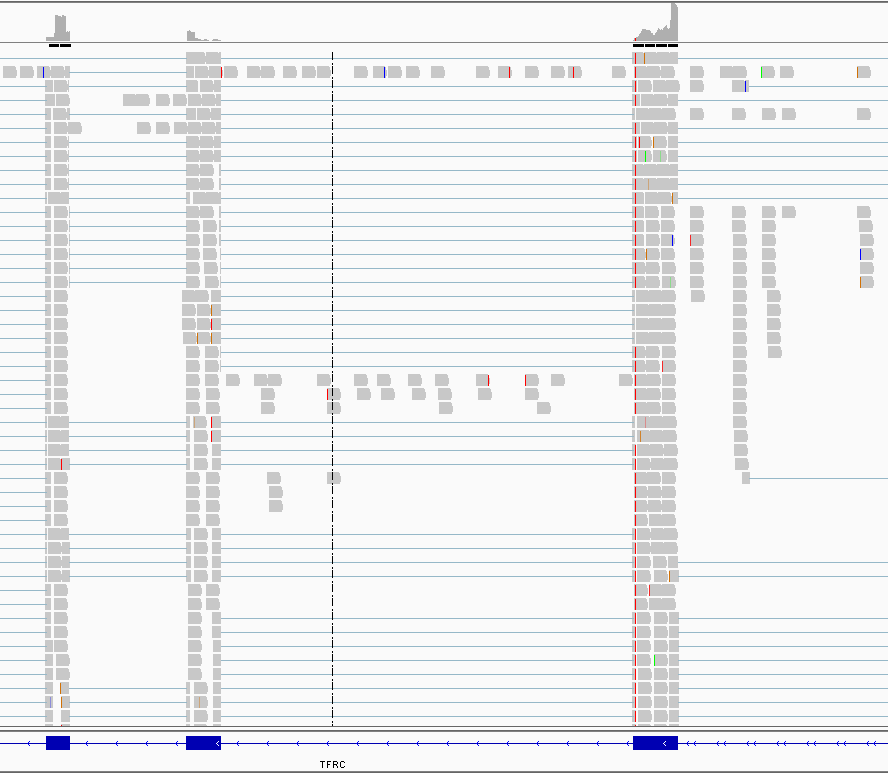
Дыло получено два файла с покрытиями. Для первой реплики их оказалось 281, а для второй на 11 меньше. Из полученного файла первой реплики были выбраны и проанализированы три гена. В файле оказалось множество трансферрин-кодирующих генов, поэтому было решено анализировать два TFRC гена.
chr3 195802030 195802231 protein_coding TFRC chr3 195801068 195801304 protein_coding TFRC chr3 195799690 195799751 snRNA RNU7-18P
Из визуализации видно, что не все ридв хорошо ложатся на гены. Некоторые расположены на интронах (~5%), а некоторые (~0.5%) ложатся на некодирующие или неаннотированные области. Это может быть связано как с ошибкой метода, так и с недостаточной изученностью экспрессии, плохим картированием.
Работа в bedtools
Обязательная часть
Задача: какие гены и насколько глубоко оказались покрыты чтениями, которые мы анализировали в выше.
Нужно описать эти гены: размер, координаты, функция, количество экзонов и интронов, направление, любая дополнительная информация.
| Кордината начала | Кордината начала | Имя | Функция | Покрытие | Направление |
| 195802030 | 195802231 | TFRC | Продукт гена необходим для доставки железа от трансферрина в клетку. Является трансмембранным гликопротеином, состоит из двух мономеров, соединённых дисульфидной связью. Каждый мономер связывает одну молекулу голотрансферина, образуя комплекс Fe-Tf-TfR | 2284 | + |
| 195801068 | 195801304 | TFRC | -//- | 2457 | - |
| 195799690 | 195799751 | RNU7-18P | Малая ядерная некодирующая РНК. Функции продукта неизвестны. | 66 | - |
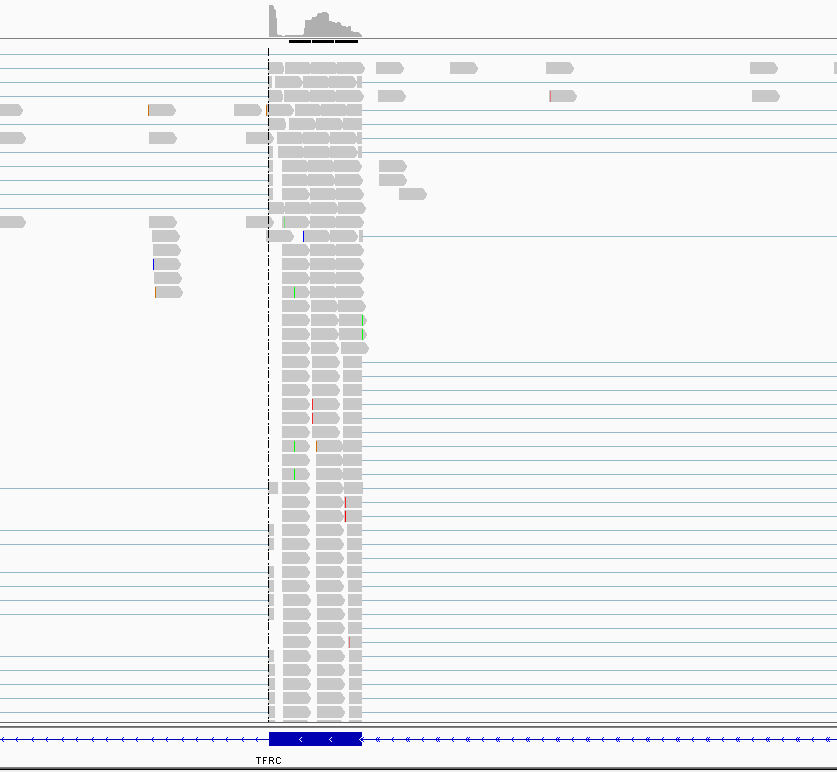
|
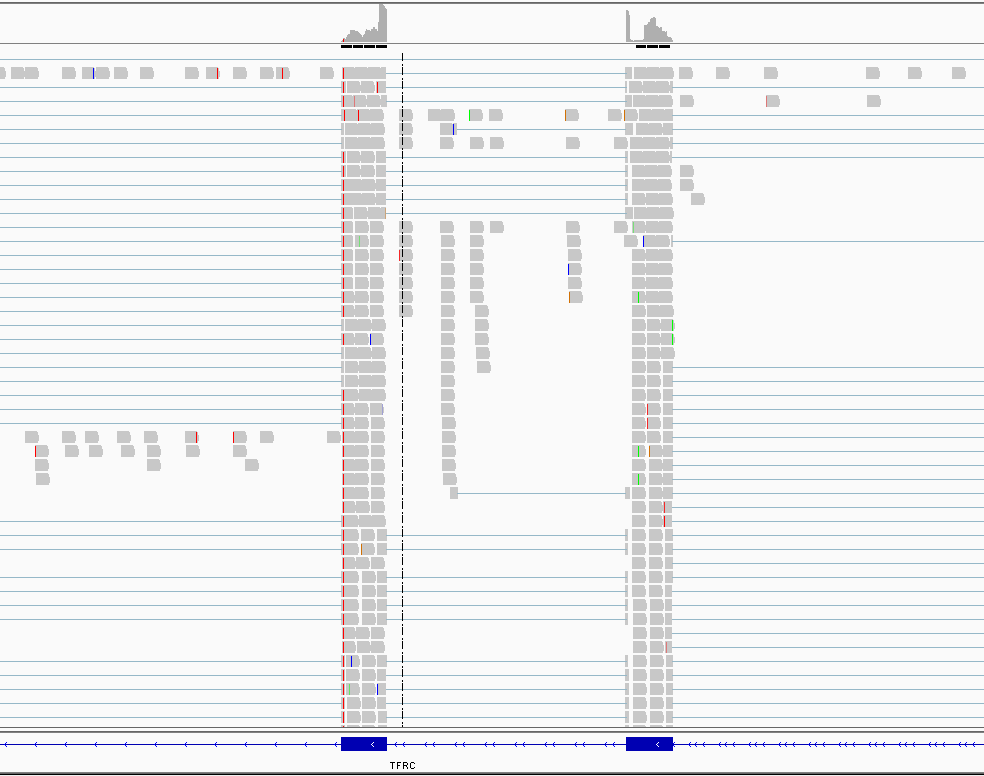
|
|
Дополнительные задания
1. Получите из файла в выравниванием файл с чтениями в формате fastq
Исходные данные: alignment_sorted1.bam
Команда: bedtools bamtofastq -i alignment_sorted1.bam -fq chr3.1_all.fq
Результат: chr3.1_all.fq
2. Получите файл с нуклеотидной последовательностью (.fasta) для одного из покрытых Вашими чтениями генов.
Для этого нужно создать файл, с которым мы будем пересекать наши чтения.
Исходные данные: chr3_my_gene.bed
Команда: bedtools getfasta -fi chr3.fasta -bed chr3_my_gene.bed > my_gene.fasta
Результат: my_gene.fasta
3. Разбейте свою хромосому на фрагменты по 1 млн нуклеотидов. Какова длина хромосомы в нуклеотидах? Сколько в результате получилось интервалов?
Исходные данные: my_chromosome_l.txt
Команда: bedtools makewindows -g my_chromosome_l.txt -w 1000000 > my_chromosome_by_1000000.txt
Результат: my_chromosome_by_1000000.txt
У меня получилось, что хромосома длиной была разбита на 200 частей.
4. Объедините Ваши чтения в кластеры (используйте bed файл с выровненными чтениями из Обязательной части задания).
Исходные данные: chr3.1_s.bed
Команда: bedtools cluster -i chr3.1_s.bed -d 7 > chr3.1_clusters.txt
Результат: chr3.1_clusters.txt
5. Наберите из Вашей хромосомы 1000 случайных фрагментов по 200 нуклеотидов.
Исходные данные: my_chromosome_l.txt, chr3.fasta
Команда1: bedtools random -g my_chromosome_l.txt -n 1000 -l 200 > random_1000_200.bed #Сгенерировали 1000 последовательностей длины 200 учитывая длину хромосомы 3.
Команда2: bedtools getfasta -fi chr3.fasta -bed random_1000_200.bed > random_parts.bed
Результат: random_parts.bed, random_1000_200.bed
6. Получите координаты 3`-области одного из покрытых Вашими чтениями генов длиной в 1000 нуклеотидов. --/ In work /--
Исходные данные:
Команда:
Результат:
7. Получите координаты одного из покрытых Вашими чтениями генов, расширенные на 1000 нуклеотидов в обе стороны.
Исходные данные: my_chromosome_l.txt, my_gene.bed
Команда: bedtools slop -i my_gene.bed -g my_chromosome_l.txt -b 1000 > my_gene_slopped.bed
Результат: my_gene_slopped.bed
8. Получите координаты одного из покрытых Вашими чтениями генов, сдвинутые на 500 нуклеотидов ближе к началу хромосомы.
Исходные данные: my_chromosome_l.txt, my_gene.bed
Команда: bedtools shift -i my_gene.bed -g my_chromosome_l.txt -s -500 > my_gene_shifted.bed
Результат: my_gene_shifted.bed
9. Получите непересекающиеся фрагменты, соответствующие области, покрытой Вашими чтениями (используйте bed файл с выровненными чтениями из Обязательной части задания). --/ In work /--
Исходные данные:
Команда:
Результат:
10. Получите файл с координатами интервалов, покрытых Вашими чтениями, с информацией о покрытии в любом формате. --/ In work /--
Исходные данные:
Команда:
Результат:
| Команда | Результат |
| fastqc chr3.fastq | Анализ качества ридов |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr3.fastq chr3_outfile.fastq TRAILING:20 MINLEN:50 | Очистка чтений |
| export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 | Прописывает временный путь до исполняемой программы |
| hisat2-build chr3.fasta chr3 | Индексирует референсную последовательность, записывает результат в *.ht2 файлы |
| hisat2 -x chr3 -U chr3_outfile.fastq --no-spliced-alignment --no-softclip > alignment.sam | Строит выранивание прочтения и референса, записывает результат в .sam файл |
| samtools view alignment.sam -b -o alignment.bam | Переводит выравнивание в бинарный формат |
| samtools sort alignment.bam -T out_sort.txt -o alignment_sorted.bam | Индексирует референсную последовательность, записывает результат в *.ht2 файлы |
| samtools index alignment_sorted.bam | Индексирует отсортированный .bam файл |
| Команда | Результат |
| export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 | Прописывает временный путь до исполняемой программы |
| hisat2-build chr3.fasta chr3 | Индексирует референсную последовательность, записывает результат в *.ht2 файлы |
| hisat2 -x chr3 -U chr3.[1/2].fastq --no-softclip > alignment[1/2].sam | Строит выранивание прочтения и референса, записывает результат в .sam файл |
| samtools view alignment[1/2].sam -b -o alignment[1/2].bam | Переводит выравнивание в бинарный формат |
| samtools sort alignment[1/2].bam -T out_sort[1/2].txt -o alignment_sorted[1/2].bam | Индексирует референсную последовательность, записывает результат в *.ht2 файлы |
| samtools index alignment_sorted[1/2].bam | Индексирует отсортированный .bam файл |
| samtools idxstats alignment_sorted[1/2].bam > out[1/2].txt | Записывает откартировавшиеся прочтения |
| bedtools bamtobed -i alignment_sorted[1/2].bam > chr3.[1/2]_s.bed | Получает на вход .bam файл, а выдаёт .bed соответственно |
| bedtools intersect -a gencode.genes.bed -b chr3.[1/2]_s.bed -u > chr3.[1/2]_crossed.bed | Ищет перекрывание множества генов генома с множеством ридов выравнивания. -u отбираются лишь гены с ненулевым покрытием. |
| bedtools bamtofastq -i alignment_sorted1.bam -fq chr3.1_all.fq | Переводит bam в fastq формат |
| bedtools getfasta -fi chr3.fasta -bed chr3_my_gene.bed > my_gene.fasta | Нарезает хромосому по разметке и кладёт каждый кусочек под свой ID |
| bedtools makewindows -g my_chromosome_l.txt -w 1000000 > my_chromosome_by_1000000.txt | Режет хромосому по рамке в 1000000 нуклеотидов |
| bedtools cluster -i chr3.1_s.bed -d 7 > chr3.1_clusters.txt | Собирает риды в кластеры, если они ближе, чем 7 нуклеотидов |
| bedtools random -g my_chromosome_l.txt -n 1000 -l 200 > random_1000_200.bed | Создаёт случайные рамки |
| bedtools getfasta -fi chr3.fasta -bed random_1000_200.bed > random_parts.bed | Получает файл с нарезанными по координатам кусочками |
| bedtools slop -i my_gene.bed -g my_chromosome_l.txt -b 1000 > my_gene_slopped.bed | Расширяет рамки на 1000 |
| bedtools shift -i my_gene.bed -g my_chromosome_l.txt -s -500 > my_gene_shifted.bed | Сдвиг рамки влево или вправо (ближе или дальше от начала\конца хромосомы) |
Ссылки
- Распределение файлов с ридами
- FastQC
- Trimmomatic
- Руководство Trimmomatic
- samtools
- bcftools
- annotate_variation.pl
- convert2annovar.pl.
- bedtools: a powerful toolset for genome arithmetic
- TFRC
- samtools
- samtools
- samtools
© Кравченко Павел
2017