EMBOSS: пакет программ для анализа последовательностей
Следите за обновлениями и дополнениями
Если Вы заметили опечатки, или ссылка испортилась, пожалуйста, напишите мне
1. Несколько файлов в формате fasta собрать в единый файл
Исходные данные: ./to_join
Команда: seqret "*.fasta" out.fasta
Результат: out.fasta
Или
Исходные данные: mylist.txt
Команда: seqret @mylist.txt out.fasta
Результат: out.fasta
2. Один файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы
Исходные данные: coding1.fasta
Команда: seqretsplit coding1.fasta
Результат: ./to_split/out
3. Из файла с хромосомой в формате .gb (была взята последовательность митохондриальной хромосомы человека) вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле
Исходные данные: mylist.txt
Команда: seqret @mylist.txt out.fasta
Результат: out.fasta
4, Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Результат - в одном fasta файле
Исходные данные: Saccharomyces_COX2.fasta
Команда: transeq Saccharomyces_COX2.fasta Saccharomyces_COX2_pep_out.fasta -table 0
Результат: Saccharomyces_COX2_pep_out.fasta
5. Транслировать данную нуклеотидную последовательность в шести рамках
Исходные данные: coding.fasta
Команда: transeq coding.fasta coding_out.fasta -table 0 -frame 6
Результат: coding_out.fasta
6. Перевести выравнивание и из fasta формате в формат .msf
Исходные данные: alignment.fasta
Команда: seqret alignment.fasta msf::alignment.msf
Результат: alignment.msf
7. Выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имя последовательности и число)
Исходные данные: alignment.fasta
Команда: infoalign alignment.fasta -refseq 2 -only -name –idcount -outfile stdout
Результат записан в файле: out_alignment.txt
8. (featcopy) Перевести аннотации особенностей в записи формата .gb в табличный формат .gff
Исходные данные: chromosome.gb
Команда: featcopy chromosome.gb chromosome.gff
Результат: chromosome.gff
9. (extractfeat) Из данного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями; (*) добавить в описание каждой последовательности функцию белка (из поля product)
Исходные данные: sequence.gb
Команда: extractfeat sequence.gb -type CDS -describe^Crodust prot_seq_out.fasta
Результат: prot_seq_out.fasta
10. Перемешать буквы в данной нуклеотидной последовательности
Исходные данные: coding.fasta
Команда: shuffleseq coding.fasta coding_shuffle.fasta
Результат: coding_shuffle.fasta
11. (*) Для случайной последовательности проверить с помощью blastn сколько "достоверных" находок (с E-value < 0.1) найдется в нуклеотидном банке данных (запустите blastn с порогом E = 10 - по умолчанию и посчитайте сколько с E-value < 0.1)
Исходные данные: -
Команда: makenucseq -amount 1 -length 100 random_blast.fasta
Результат: random_blast.fasta Была найдена всего 1 находка с E-vale > 4
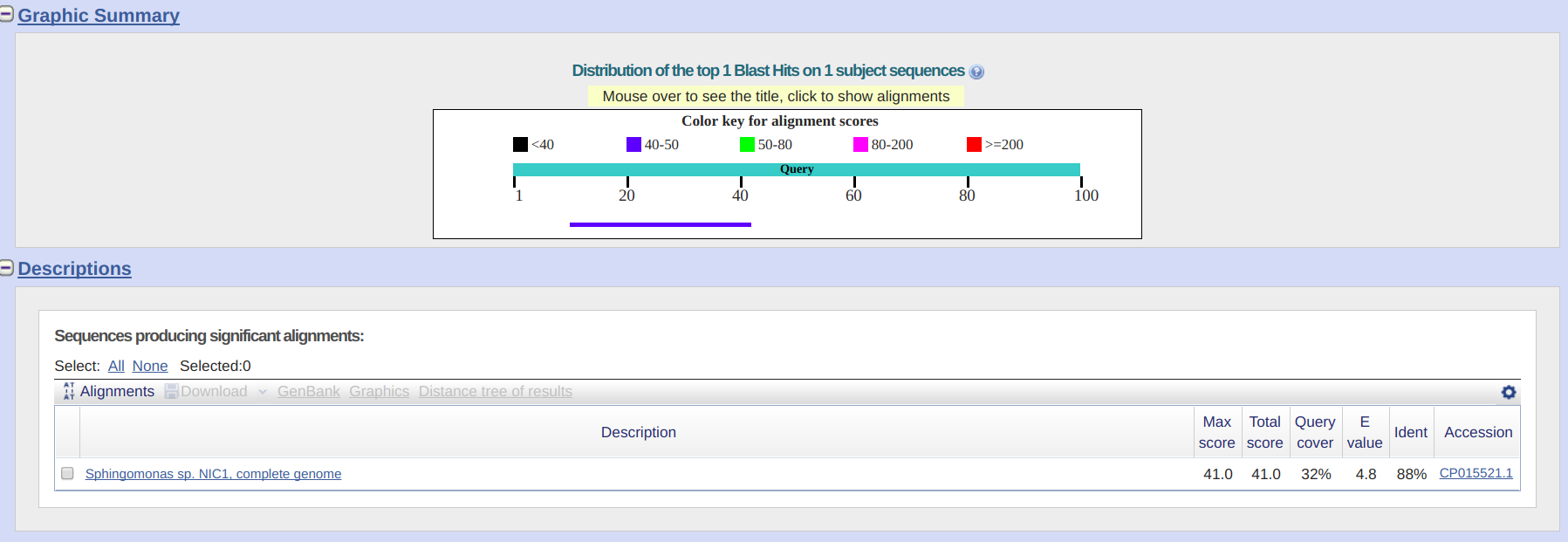
|
13. Найдите частоты кодонов в данных кодирующих последовательностях
Исходные данные: coding2.fasta
Команда: cusp coding2.fasta coding2_out.fasta
Результат: coding2_out.fasta
15. (tranalign) Выровняйте кодирующие последовательности соответственно выравниванию белков - их продуктов
Исходные данные: Белковое выравнивание, выполненное c помощью команды needle Cyanea_capillata_CO1_pep_al.fasta и файл с нуклеотидной последовательностью
Cyanea_capillata_CO1.fasta
Команда: tranalign -aseq Cyanea_capillata_CO1.fasta -bseq Cyanea_capillata_CO1_pep_al.fasta -outseq Cyanea_capillata_CO1_out.fasta
Результат: Cyanea_capillata_CO1_out.fasta
При выполнении команды возникает ошибка.
Error: Guide protein sequence KM281976.1_1 not found in nucleic sequence KM281976.1_1
Error: Guide protein sequence KM281983.1_1 not found in nucleic sequence KM281983.1_1
Warning: No sequences written to output file 'Cyanea_capillata_CO1_out.fasta'
16. Постройте локальное множественное выравнивание трех нуклеотидных последовательностей
Исходные данные: ./to_split/3
Команда: edialign @my_list.txt -outfile out_16.fasta -outseq out.fasta
Результат: out_16.fasta out.fasta
17. Удалите символы гэпов и другие посторонние символы из последовательности
Исходные данные: coding_gap.fasta
Команда: degapseq coding_gap.fasta coding_gap_out.fasta
Результат: coding_gap_out.fasta
18. Переведите символы конца строки в формат unix
Исходные данные: Mouse_Mus_cytb.fasta
Команда: noreturn Mouse_Mus_cytb.fasta Mouse_Mus_cytb_unix.fasta
Результат: Mouse_Mus_cytb_unix.fasta
19. Создайте три случайных нуклеотидных последовательностей длины сто
Исходные данные: -
Команда: makenucseq -amount 3 -length 100 random_100.fasta
Результат: random_100.fasta
20. Файл с ридами sra_data.fastq в формате fastq перевести в формат fasta
Исходные данные: sra_data.fastq
Команда: seqret sra_data.fastq fasta::sra_data.fasta
Результат: sra_data.fasta
© Кравченко Павел
2017