Поиск сайта связывания транскрипционного фактора, регулирующего синтез пуринов у гаммапротеобактерии Haemophilus influenzae , с помощью программы MEME
Следите за обновлениями и дополнениями
Если Вы заметили опечатки, или ссылка испортилась, пожалуйста, напишите мне
В данном практикуме был проанализирован путь биосинтеза пуринов у гаммапротеобактерии Haemophilus influenzae .
Поиск сайта связывания транскрипционного фактора, регулирующего данный процесс был произведён с помощью программы MEME.
Для изучаемой бактерии известно 295 последовательностей белков, участвующих в биосинтезе пуринов (Поиск по базе данных UniProtKB
keyword:"Purine biosynthesis [KW-0658]" organism:"haemophilus influenzae"). Из них 34 были проверены специалистами, и имели статус Reviewed.
Список Popular organisms для данной бактерии состоял из: HAEIN (16), HAEI8 (6), HAEIE (6), HAEIG (6).
|
|
Таблица 2 содержит в себе информацию о идентификаторах записей белков, UniprotKB ID, именах генов, кодирующих белки.
|
AC записи EMBL, описывающей геном: CP020006.1. Для данного штама организма по приведённому идентификатору был скачан анотированный геном и геном в fasta формате.
Из второго средствами командной строки Linux и пакета EMBOSS (seqret extractfeat, descseq, extractseq) были извлечены upstream области генов длиной 100 нуклеотидов. Учитывалась прямая и обратная цепь.
Найденные области были записаны в
файл для дальнейшего анализа.
Ссылка на скрипт
Был выполнен анализ последовательностей с помощью программы ememe из пакета EMBOSS.
С помощью команды
ememe -dataset ./files/out.fasta -outdir . -nmotifs 3 -revcomp Y
был получен отчёт в html формате, некоторые моменты из которого обсуждаются ниже.
Как мы и задали, было найдено три мотива. Для каждой находки программа выдаёт LOGO диаграмму, отражающую вероятность встречи определённого нуклеотида в конкретной позиции мотива.
Также LOGO содержит дополнительную информацию о информационном содержании данного столбца выравнивания.
Мотив 1
width = 10 sites = 7 llr = 80 E-value = 1.0e-003
На рисунке 2 показано выравнивание с сайтами, взятыми в мотив. Видно, что семь последовательностей входит в первый мотив с хорошим p-value, что может говорить о высокой вероятности значимости полученного результата. В целом, мотив имеет низкое значение E-value, что делает находку правдоподобной. Также она содержит всего 7/8 последовательностей.
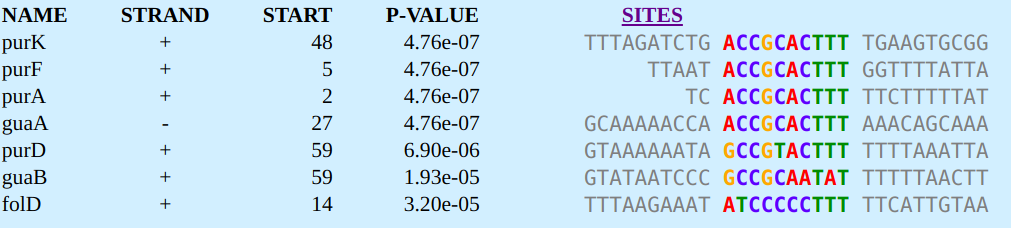 |
Мотив 2
width = 9 sites = 8 llr = 67 E-value = 9.0e+002
Во второй мотив вошло уже восемь последовательностей; видно, что они имеют по-отдельности p-value как меньше, так и больше последовательностей в первом мотве.
Данный мотив имеет высокое значение E-value, что говорит о его низкой превдоподобности.
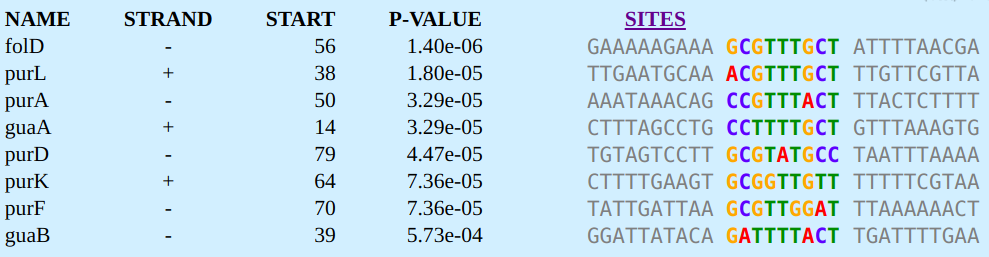 |
Мотив 3
width = 8 sites = 2 llr = 23 E-value = 4.2e+003
В третий мотив входит две последовательности. Данный результат по надёжности, пожалуй, самый неправдоподобный. Значение E-value так же очень большое.
Доверия к такому мотиву нет.
 |
Программа выдаёт также SUMMARY OF MOTIFS, в котором отражены позиции найденных мотивов в рассматриваемом нами фрагменте upstream области. В целом, вервый мотив можно трактовать как надёжный.
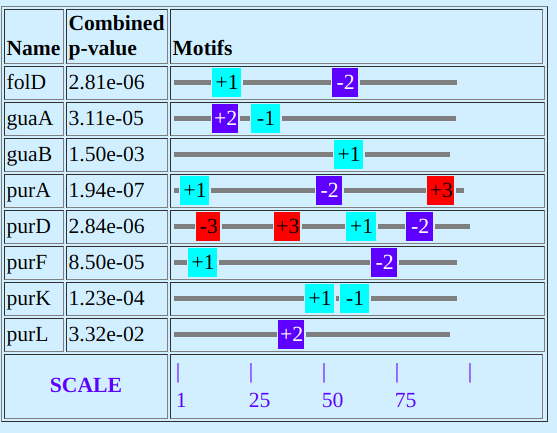 |
Найдите в Интернете LOGO для сайта связывания пуринового репрессора E.coli и сравните его с LOGO вашего мотива (мотивов).
Было найдено несколько LOGO диаграмм:- The PurR regulon in Escherichia coli K-12 MG1655

Рисунок 8. E. Coli LOGO
- Involvement of the ribose operon repressor RbsR in regulation of purine nucleotide synthesis in Escherichia coli
Возможно, данный мотив сходен с найденным при выполнении практикума.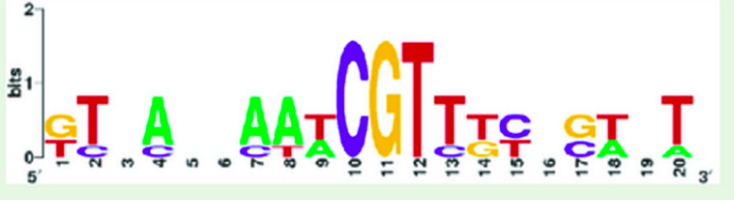
Рисунок 9. E. Coli LOGO
- Computer analysis of transcription regulatory patterns in completely sequenced bacterial genomes
Рисунок 10. E. Coli LOGO
Диаграммы в статьях заметно отличаются от найденных в нашей работе. Так может получиться из-за различия в последовательностях между рассматриваемыми организмами, малой длины наших последовательностей, их малой численности.
Проведите программой emast поиск найденных мотивов в полном геноме вашей бактерии.
Был проведён анализ с помощью программы meme.emast заданной последовательности. С результатом работы программы вы можете ознакомиться по ссылке ниже.Отчёт
DATABASE ./CP020006.fasta (nucleotide) Last updated on Fri Mar 30 00:00:11 2018 Database contains 1 sequences, 1857175 residues Scores for positive and reverse complement strands are combined. MOTIFS ./meme.txt (nucleotide) MOTIF WIDTH BEST POSSIBLE MATCH ----- ----- ------------------- 1 13 GCAAAAGAGAGCG 2 18 AAAGTGCGGCTGTTTTTT 3 13 CCCCCTAGCCCCC PAIRWISE MOTIF CORRELATIONS: MOTIF 1 2 ----- ----- ----- 2 0.30 3 0.23 0.09 No overly similar pairs (correlation > 0.60) found. Random model letter frequencies (from non-redundant database): A 0.274 C 0.225 G 0.225 T 0.274Видно, что находки скудны. Мы нашли всего одно хорошее совпадение, которое, впрочем, может быть лишь совпаденеим. Аннотация последовательностей затруднительна на выбранном организме ио с выбранным объектом.
Ссылки
- Uniprot
- EMBL
- extractseq
- descseq
- extractfeat
- Computer analysis of transcription regulatory patterns in completely sequenced bacterial genomes
© Кравченко Павел
2018