Практикум 12. Алгоритмы и программы множественного выравнивания
2. Сравнение выравниваний одних и тех же последовательностей тремя разными программами
Для сравнения выравниваний было выбрано семейство доменов PF00025 ADP - ribosylation factor family (из PFAM).
Выравнивания производились сервисами, доступными в Jalview (A - Mafft, B - Muscle, C - ClustalO).
Файл выравнивания, произведенного программой Mafft .
Файл выравнивания, произведенного программой Muscle .
Файл выравнивания, произведенного программой ClustalO .
Проект Jalview со сравниваемыми выравниваниями.
Попарное сравнение производилось ручным методом в Jalview. Результаты сравнения выравниваний представлены ниже.
Таблица 1. Сравнение выравниваний программами Mafft и Muscle.
Таблица 2. Сравнение выравниваний программами Mafft и ClustalO.
Наибольшее сходство удалось увидеть в паре сравнения Mafft - ClustalO. Практически все блоки совпадают по позициям.
3. Построение выравнивания по совмещению структур и сравнение его с выравниванием программой MSA
Для проведения выравнивания были выбраны белки с доменом PF00089:
1ae5:A - HUMAN HEPARIN BINDING PROTEIN
1c1q:A - RECRUITING ZINC TO MEDIATE POTENT SPECIFIC INHIBITION OF SERINE PROTEASES
1dpo:A - STRUCTURE OF RAT TRYPSIN
1. Сначала было произведено выравнивание последовательностей, полученное из совмещения структур, программой PDBeFold. Результат выравнивания в формате fasta представлен в файле . Визуализация совмещения произведена на сайте PyMol (рис.1)
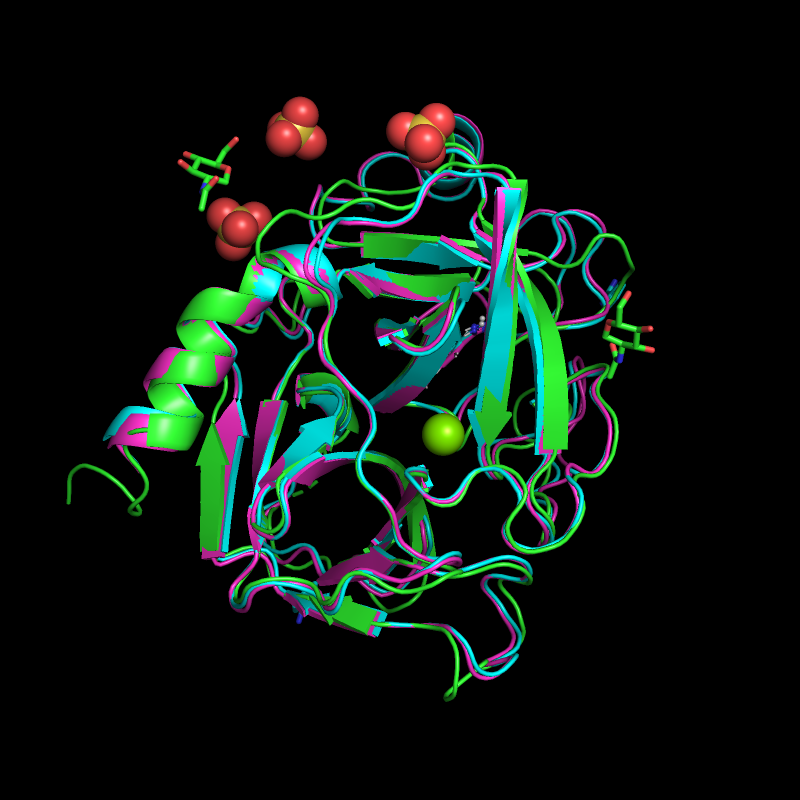
2. Затем было произведено выравнивание последовательностей программой Mafft (MSA). Результат выравнивания в формате fasta представлен в файле .
3. Cравнение выравниваний производилось ручным методом в Jalview. С проектом Jalview со сравниваемыми выравниваниями можно ознакомиться по ссылке Jalview . Результаты сравнения выравниваний представлены ниже.
Таблица 3. Сравнение выравниваний .
Проанализировав полученные данные из таблицы 3, можно сделать вывод, что схожесть данных выравниваний достаточно большая. Процент совпадающих колонок для выравнивания с помощью совмещения пространственных структур и с помощью программы множественного выравнивания MAFFT велик.
4. Программа CLUSTALW (MSA)
ClustalW — это широко используемая программа для множественного выравнивания последовательностей. В последнее время она активно применяется для построения множественных выравниваний последовательностей во многих рецензируемых научных статьях. В этой программе используется прогрессивный метод построения выравниваний: вместо одновременного выравнивания всех последовательностей она добавляет их по одной. W в ClustalW обозначает веса, потому что программа использует сложную схему, позволяющую каждой последовательности получать пропорциональный вес, чтобы очень похожие последовательности в конечном итоге не доминировали при выравнивании нескольких последовательностей. [1]
ClustalW осуществляет три этапа алгоритма:
1. Парное выравнивание и расчёт матрицы расстояний.
Для всех пар последовательностей выполняется попарное выравнивание, результаты которого используются для построения матрицы расстояний, учитывающей дивергенцию последовательностей.
2. Построение направляющего (филогенетического) дерева.
На основе матрицы расстояний с помощью метода neighbour-joining (присоединения соседей) формируется дерево, которое определяет порядок добавления последовательностей при выравнивании.
3. Прогрессивное выравнивание.
Последовательности выравниваются в соответствии с порядком ветвей в направляющем дереве — от кончиков к корню. На каждом этапе применяется полное динамическое программирование с использованием матрицы оценок.[2]
Особенности программы ClustalW:
1. Весовые коэффициенты последовательностей.
При частичном выравнивании каждой последовательности присваиваются индивидуальные веса, чтобы снизить влияние почти повторяющихся последовательностей и увеличить вес наиболее расходящихся.
2. Штрафы за пробелы.
Используются штрафы за пробелы, специфичные для остатков, а также локально уменьшенные штрафы в гидрофильных областях, что стимулирует открытие новых пробелов в областях потенциальных петель.
3. Ограничения.
По умолчанию ClustalW рассчитан на работу с небольшим количеством последовательностей (до 500) или с файлами размером до 10 МБ. При больших объёмах данных процесс может занимать много времени или не завершаться вовсе.
4. Форматы ввода.
Программа поддерживает несколько форматов данных, включая FASTA, Swiss-Prot, PIR и другие.[1]
5. Источники
1. K. Chaichoompu, Surin Kittitornkun - King Mongkut's Institute of Technology Ladkrabang, Sissades Tongsima - National Center for Genetic Engineering and Biotechnology. MT-ClustalW: multithreading multiple sequence alignment, May 2006, DOI:10.1109/IPDPS.2006.1639537.
2. Выравнивание нуклеотидных и белковых последовательностей с помощью ClustalW (Электронный ресурс). - Режим доступа: https://clustalw.com/