Практикум 12.
Задание 1. Сравнение выравниваний Muscle и MAFFT.
Для сравнения выравнивания разными программами были выбраны следующие последовательности:
- AAT_ECOLI
- AAT_THEMA
- AAT_RHIME
- AAPAT_THET8
- AATC_YEAST
Далее в Jalview были сделаны выравнивания с помощью программ muscle (ссылка на выравнивание) и mafft (ссылка на выравнивание). Я сравнил эти выравнивания, используя прграмму Лизы Плешко.
Команда: python compare_alig.py -a1 muscle.fa -a2 mafft.fa -o res.csv
Вывод программы:
- Доля одинаково выравненных позиций в первом выравнивании: 0.64%
- Доля одинаково выравненных позиций в втором выравнивании: 0.62%
Ссылка на файловую выдачу программы.
Совпадающие участки:
| Muscle | 18-41 | 62-181 | 196-205 | 223-232 | 257-276 | 286-327 | 341-358 | 364-374 | 407-415 | 429-451 |
|---|---|---|---|---|---|---|---|---|---|---|
| MAFFT | 18-41 | 64-183 | 198-207 | 228-237 | 264-283 | 294-335 | 349-366 | 373-383 | 416-424 | 442-464 |
При просмотре двух выравниваний обнаружилось, что в несовпадаюших участках есть большое количество гэпов и различных неидентичных аминокислот. Основываясь на это, можно сделать преположение, что несоответсвия в выравниваниях вызваны различиями в алгоритмах работы программ, так как в основном не совпадалили участки с гэпами (то есть те учаски, которые вряд ли являются консервативными, а потому не подлежат точному выравниванию).
Задание 2. Сравнение выравнивания по совмещению структур с выравниванием MSA.
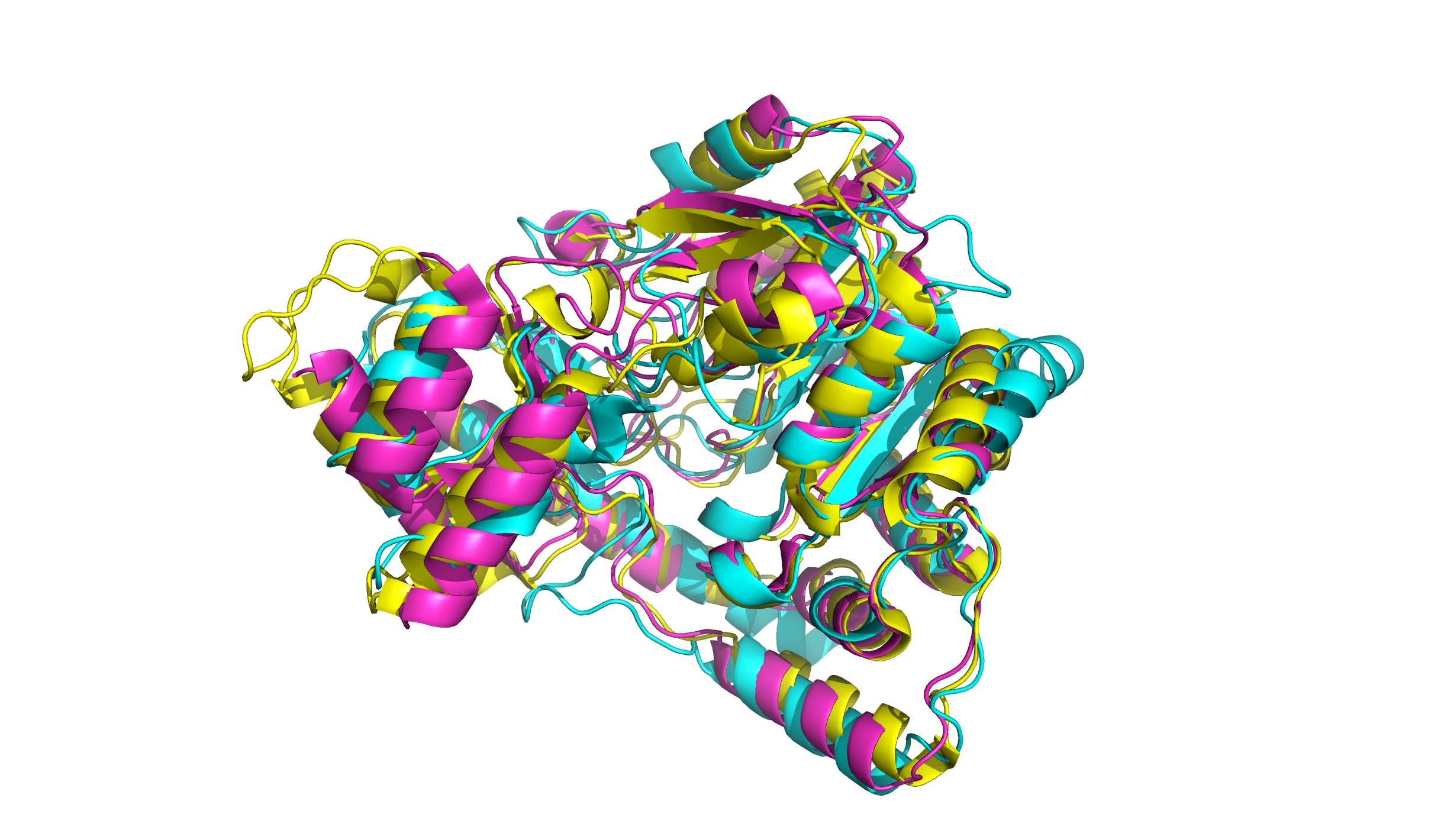
Для данного задания были выбраны первые три белка из задания 1. Выравнивание структур было сделано в Pymol с помощью команды align. На приведенном ниже изображении хорошо видно гомологию данных белков. Далее в Jalview было сделано выравнивание последовательностей данных белков. В нем присутствуют множество идентичных участков, что также сведетельтвует об общности их происхождения.
Задание 3. Описание прграммы Muscle.
Muscle использует две величины: k mer (для не выровненных пар) и Kumira (для выровненных).В данной программе можно выделить три этапа:
Этап 1: на данном этапе задача программы с максимальной скоростью (пренебрегая точностью) создать множественное выравнивание. Для каждой пары последовательностей вычисляется расстояние k-mer; из этих данных создается матрица расстояний D1, на основе которой строится двоичное деревно TREE1 с помощью алгоритма класторизации UPGMA. Далее происходит последовательное выравнивание так, что для каждого узла строится его профиль на основе попарного выравнивания профилей его дочерних узлов (построение профилей начинается с вершин дерева). В конце первого этапа мы получаем выравнивание MSA1
Этап 2: после создания TREE1 происходит его переоценка для повышения точности. Программа создает матрицу D2 на основе вычисленных расстояний Kumira, которые являются более точным чем расстояния k mer. После аналогичным первому этапу образом создается дерево TREE2, однако новое выравнивание MSA2 вычисляется только для ветвей, порядок ветвления которых изменился относительно TREE1.
Этап 3: на третьем этапе происходит доработка выравнивания MSA2. Программа, начиная с вершин дерева, разделяет TREE2 на два поддерева путем удаления выбранного ранее ребра. Даллее вычисляется профиль множественного выравнивания для каждого из них по алгоритму описанному в этапе 1. Если значение SP (величина, соответствующая доли совпадающих пар аминокислот в выравнивании) улучшается, новое выравнивание сохраняется, в противном случае оно отбрасывается. Данная оперция повторяется, пока не будет достигнут установленный ползователем предел или сходимость.
Ссылка на статью, на основе которой было сделано описание программы Muscle.