BLAST
Задание 1.
Открыв страцу BLAST на сайте NCBI, я зарегистрировался в системе. Затем открыл Protein Blast,
выбрал алгоритм blastp, выбрал базу данных Refseq_protein и нажал на кнопку "BLAST".
В ответ BLAST выдал мне 100 последовательностей парных выравниваний с ними. При этом
все найденные последовательности принадлежали бактериям. Однако E-value наихудшей
находки равен 1*10^(-38). Следовательно, в параметрах поиска надо увеличить количество
выдаваемых результатов. Онако, когда я выбрал количество выдаваемых результатов до 1000,
самый максимальный E-value был равен 2*10^(-25). Тогда я выбрал максимум выдачи в
5000 результатов. Однако и тогда E-value был равен 1* 10^(-14).При выставлении
максимума выдачи в 10000 результатов максимальный E-value составляет 0,015. Все
остальные параметры поиска я оставил без изменений.
При таких условиях поиска BLAST выдал мне 10000 результатов, среди которых
были белки бактерий (580 видов), архей (71 вид), грибов (1 вид - Candida tropicalis MYA-3404).
При этом в выдаче было 965 находок для бактерий, 75 - архей и 1 для грибов.
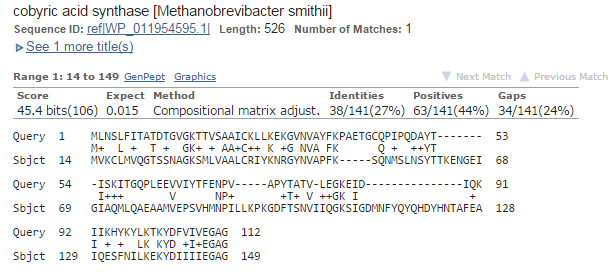
Результат наихудшей находки:
| Длина выравнивания |
136 |
| Bit score |
45,4 |
| % идентичных и сходных остатков |
Идентичных - 27%, сходных - 44% |
| E-value |
0,015 |
Картинка выравнивания:
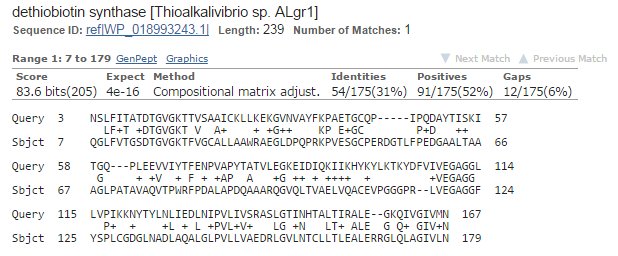
Результат выравнивания из середины списка:
| Длина выравнивания |
173 |
| Bit score |
83,6 |
| % идентичных и сходных остатков |
Идентичных - 31%, сходных - 52% |
| E-value |
4e-16 |
Картинка выравнивания:
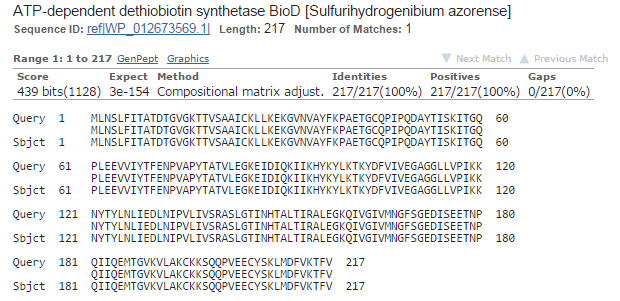
Результат лучшего выравнивания:
| Длина выравнивания |
217 |
| Bit score |
439 |
| % идентичных и сходных остатков |
Идентичных - 100%, сходных - 100% |
| E-value |
3e-154 |
Картинка выравнивания:
Далее передо мной встала задача из всей этой массы последовательностей
выделить гомологи моего белка. Гомологами я считал последовательности, чьи
E-value были меньше 1e-3 и для кторых не менее 70% запроса
вошло в полученное выравнивание (Query cover). К слову, сразу видно, что худшая
последовательность, для которой я привёл характеристики выше, гомологом исходной
последовательности не является.
Итак, для нахождения гомологов я вернулся на страницу запроса, и помимо
максимума выдачи 10000 проставил значение Expect treshold 1e-3, после чего нажал
кнопку "BLAST". Результатом стали 8176 последовательностей, среди которых, однако,
было 4 результата с Query cover < 70% при E-value 1е-3. Отметив эти
последовательности флажками, я решил ознакомиться с ними детальней, и нажал
кнопку GenPept. Два результата из этих четырёх, которые соответствовали белку
детибиотинсинтетазе, я оставил в списке гомологов, результат, соответствующий
фосфотрансацетилазе, я исключил, а результат, соответствующий белку
onanonoxo-7-onima-8-eninoihtemlysoneda, я оставил, поскольку, согласно Uniprot,
этот белок обладает детибиотинсинтетазной активностью. Получилось 8175 гомологов.
Задание 2.
В этом задании необходимо было провести поиск внутри отдельного таксона.
Вернувшись на страницу запроса, я в поле "Organism" ввёл "Escherichia coli",
выставил максимум выдачи 100 и нажал "BLAST". Среди результатов запроса выбрал
запись "ATP-dependent dethiobiotin synthetase BioD 1 [Escherichia coli]" начал
сравнивать параметры этой записи при разных поисках. Вопрос о наличии этого
результата в первом поиске даже не стоял - ведь первый поиск был внутри всей
базы данных Refseq, тогда как во втором поиске я поставил условие - искать внутри
определённого таксона. То есть, первый поиск должен в своём составе содержать
второй. Score, Query cover и %Ident результатов в обоих запросах остаётся
неизменным, в то время как E-value различаются, составляя 4е-24 в первом запросе
и 9е-26 во втором. Поскольку E-value указывает случайность находки в определённом
объёме данных, мы видим, что в первом поиске этот результат был более случаен,
чем во втором.
Задание 3.
Для выполнения этого задания я решил выбрать белок детибиотинсинтазу,
полученную из бактерии Bacillus amyloliquefaciens (запись "dethiobiotin
synthetase [Bacillus amyloliquefaciens]", Refseq AC: WP_045505834). Затем я
вернулся в меню запроса, поставил флажок "Align two or more sequences" и ввёл
в появившееся поле Refseq AC выбранного белка и нажал на "BLAST".
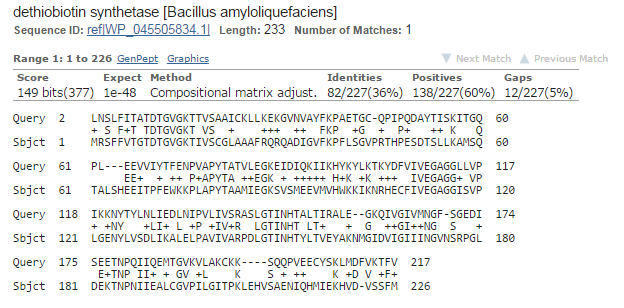
Карта локального сходства:
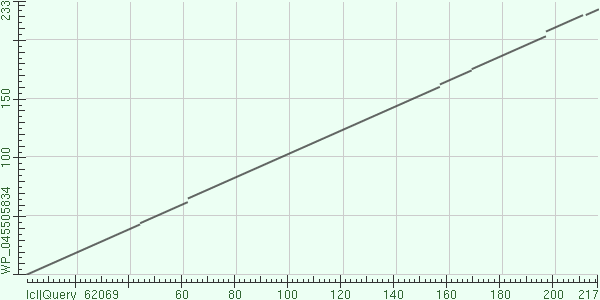
По горизонтали отложены номер аминокислотных остатков моего белка, по
вертикали - белка, с которым я сравнивал свой. По карте видно, что в выравнивании
6 открытий гэпов, что выравнивание начинается со второго аминокислотного
остатка моего белка и с первого - сравниваемого. То же можно увидеть, взглянув
на само выравнивание.
Выравнивание:
Задание 4.
Для выполнения этого задания я выбрал файл
align_03_1.fasta, взяв файд с выравниваниями
align_03.fasta из практикума 8, копировав в новый
файл его содержимое и удалив в нём гэпы. Затем я на сервере kodomo проиндексировал
получившуюся маленькую базу данных командой
makeblastdb -dbtype prot -in align_03_1.fasta -out new_database . После я
запустил в ней поиск моего бедка командой
blastp -db new_database -query YP_002729488.fasta . Лучшее из получившихся
выравниваний можно увидеть на картинке ниже.
Выравнивание в новой базе данных:
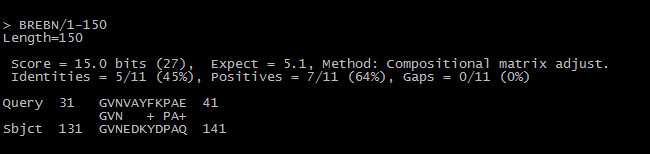
Результаты поиска в новой БД:
| Выравнивание |
Длина выравнивания |
Score |
% Identities |
% Positives |
E-value |
| BREBN |
11 |
15.0 bits |
45% |
64% |
5.1 |
Казалось бы, высокий процент совпадений, отличное выравнивание! Но тот
факт, что сама длина выравнивания очень мала - всего 11 аминокислотных остатков,
низкое значение Score относительно поиска в BLAST и очень высокий E-value
позволяют усомниться в этом утверждении. E-value, равный 5.1, указывает на
случайность совпадения, говорить о гомологии белков нельзя. Можно лишь
предположить гомологию УЧАСТКОВ этих белков, исходя из которой можно сделать
вывод, что эти участки могут выполнять сходную функцию в обоих белках.
Главная страница
© Головачев Ярослав