Ресеквенирование. Поиск полиморфизмов у человека.
Задание 1.
Для данной работы мне была дана 22-я хромосома человека (последовательность
хромосомы в fasta-формате; файл с одноконцевыми чтениями формата fastq).
Запустив команду fastqc chr22.fastq я получил архив и
файл в html-формате, визуализирующий информацию о чтениях.
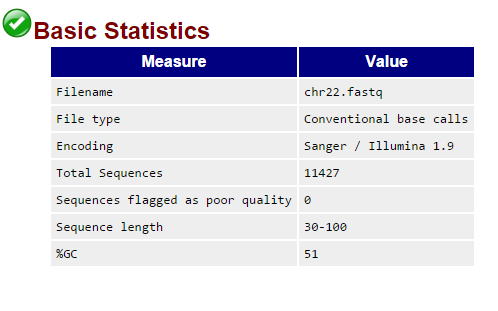
Общая информация о чтениях
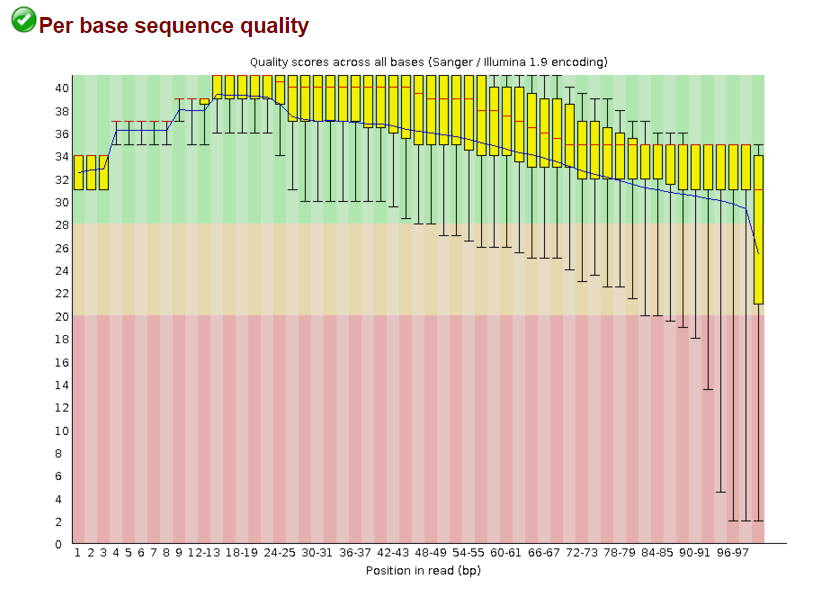
Визуализация информации о чтениях
Как видно, конец ридов прочитан хуже основной части. Это может быть связано с тем, что при увеличении
количества итераций идёт накопление ошибок.
Задание 2.
Теперь было необходимо произвести очистку чтений с помощью программы Trimmomatic. Командой
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr22.fastq out.fastq TRAILING:20 MINLEN:50
отрезал с конца каждого чтения нуклеотиды качеством ниже 20, оставив чтения длиной не менее 50 нуклеотидов.
Результат - файл out.fastq
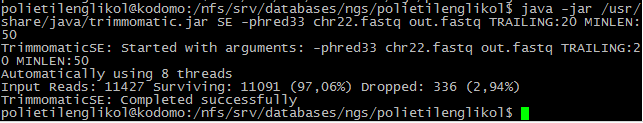
Очистка программой Trimmomatic
Программа Trimmomatic удалила 336 (2,94%) чтений.
Затем я вновь запустил FastQC.
FastQC после очистки
Результат - архив out_fastqc.zip и
html-файл с визуализацией информации о чтениях.
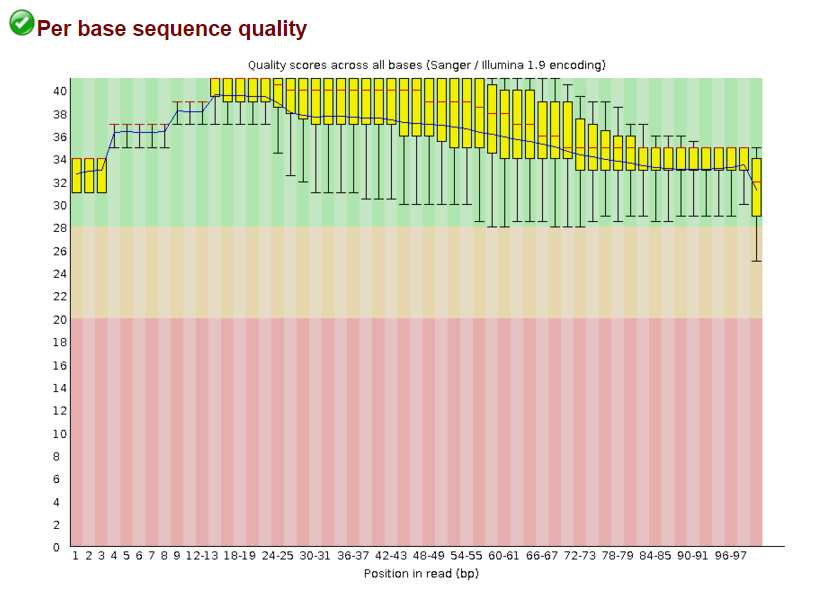
Визуализация информации о чтениях после очистки
Видно, что исчезли риды, у которых на концах были нуклеотиды плохого качества. Лишь последняя позиция
расположена в жёлтой области.
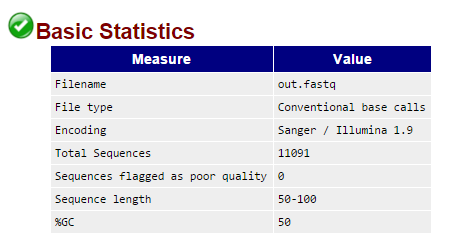
Базовая информация о чтениях (после очистки)
Как видно из данного скриншота, программа Trimmomatic оставила лишь те риды, длина которых была не
менее 50.
Задание 3.
В данном задании было необходимо откартировать уже очищенные чтения с помощью программы BWA. Для этого
я сначала проиндексировал референсную последовательность 22-й хромосомы.
Программа bwa index
Затем мной было построено выравнивание прочтений и референса:
Построение выравнивания программой bwa mem
Задание 4.
В этом задании надо было проанализировать полученное в прошлом задании выравнивание. Для начала файл
с выравниванием был переведён в бинарный формат .bam .

Затем выравнивание референса и чтений было отсортировано по координате в референсе начала чтения.

Результат - файл с отсортированными ридами (в бинарном формате
.bam).
После этого отсортированный файл был проиндексирован:

Затем была получена информация о количестве ридов, откартировавшихся на геном.

Как видно, откартировалось 11090 ридов.
Задание 5.
В данном задании было необхлдимо найти SNP и индели для их последующего анализа.
Сперва был создан файл с полиморфизмами (в формате .bcf).

Затем был создан файл со списком отличий между референсом и чтениями в формате (.vcf).

Первые же три полиморфизма из файла diff.vcf были мною описаны:
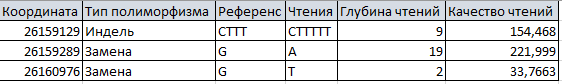
Качество и покрытие у первых двух полиморфизмов довольно неплохое, а вот у третьего - не очень.
Задание 6.
Затем было необходимо проанализировать полученные snp с помощью программы annovar (индели не
анализировались - для этого я их просто удалил), пользуясь базами данных refgene, dbsnp, 1000 genomes, GWAS, Clinvar. Для этого надо было
получить из .vcf файла другой файл, с которым будет работать annovar, с помощью скрипта convert2annovar.pl
(расположение скрипта на kodomo - /nfs/srv/databases/annovar), после чего аннотировать по базам данных
с помощью скрипта annotate_variation.pl (лежит там же).
Правда, скрипты просто так работать не стали, пришлось изменить права (команда
chmod agu+xwr convert2annovar.pl, для другого скрипта - аналогично).

Конвертация .vcf-файла в .avinput-файл
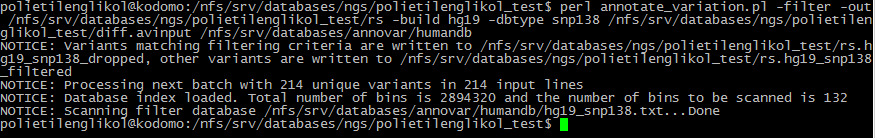
Аннотация SNP из .avinput-файла по БД dbsnp (snp138), аналогичными командами SNP были аннотированы
по другим БД
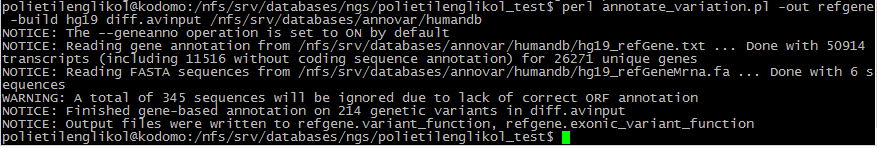
Аннотация SNP из .avinput-файла по БД refgene
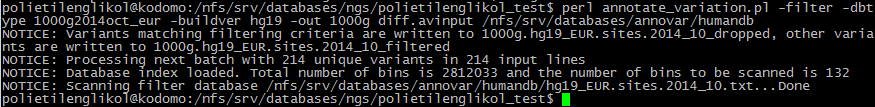
Аннотация SNP из .avinput-файла по БД 1000 genomes
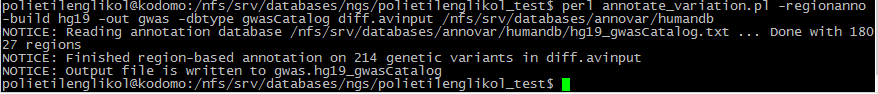
Аннотация SNP из .avinput-файла по БД GWAS
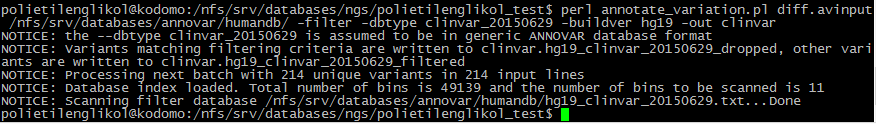
Аннотация SNP из .avinput-файла по БД Clinvar
Из полученных SNP 176 имеют rs, а 38 - не имеют (файлы rs.hg19_snp138_dropped
и rs.hg19_snp138_filtered соответственно).
При аннотировании с помощью базы RefGene было найдено 75 het и 139 hom замен. В самой БД refgene
snp делятся на несколько категорий:
- exonic - кодирующий участок
- intronic - некодирующий участок
- splicing - полиморфизм, входящий в 2-bp, участвующие в сплайсинге
- UTR5 - элемент внутри 5'-нетранслируемого региона
- UTR3 - элемент внутри 3'-нетранслируемого региона
- ncRNA - элемент внутри некодирующего транскрипта (транскрипта без аннотации в описаниии гена)
- upstream - участок в пределах 1kb после старт-кодона
- downstream - участок в пределах 1kb перед стоп-кодоном
- intergenic - межгенная область
Refgene выделил 26 exonic, 6 ncRNA_intronic, 1 ncRNA_exonic и 181 intronic полиморфизмов.
SNP попали в 3 гена:
- MYO18B - ген миозина XVIIIB, участвующего в регуляции мышечных генов
- TTC28 - ген белка tetratricopeptide repeat protein 28, участвующего в образовании веретена деления
- APOL1 - ген аполипопротеина L1, участвующего в транспорте липидов по телу
Стоит отметить, что полиморфизмы, попавшие в ncRNA, входили в состав гена TTC28, и выдаче Refgene
отмечались как TTC28-AS1 - то есть, TTC28 antisense RNA 1. Также стоит сказать, что экзоны были затронуты
в каждом из генов.
Информацию по покрытию и качеству полиморфизмов можно взять в аннотациях по любой БД, но больше всего
аннотаций было выдано по БД Refgene, поэтому информацию я брал оттуда. Аннотации с хорошим покрытием также
имеюли высокое качество чтений. Аннотаций с покрытием не более 4 было 130 штук, не менее 5 (довольно хорошее) - все остальные
(84). 72 аннотации имели покрытие выше 10.
Информация по аминокислотным заменам, к которым привели SNP, хранится в файле
refgene.exonic_variant_function. Всего замен получилось
126, из них 18 несинонимичных и 8 синонимичных.
Информацию о частоте аллели я брал из аннотаций БД 1000genomes (данные нашлись для 140 аннотаций).
Довольно много попалось редких полиморфизмов (с частотой от 0,001 до 0,3) - 66 штук, остальные 74
полиморфизма довольно распространены (частота > 0,3).
Клиническая аннотация полиморфизмов приведена в БД Gwas. В 22-й хромосоме было найдено 3 полиморфизма
по этой БД. Первый полиморфизм - в гене MYO18B, охарактеризован как отвечающий за нарушения математических
способностей у детей с дислексией. Второй - в гене TTC28, ожирение. Третий полиморфизм - в гене APOL1,
гломерулосклероз.
Таблица с выдачей аннотаций по разным БД.
Ссылка на главную страницу
© Головачев Ярослав