Задание 1 |
| Для данного задания был выбрана структура 1DUP. Гомологи были найдены при помощи поиска по сходству структур в PDBeFold. |
| Были найдены структурные гомологи для цепи А с RMSD между 0,8 и 2,5 и длиной выравнивания более 50% от длины 1DUP. Выбранные гомологи представлены в таблице. |
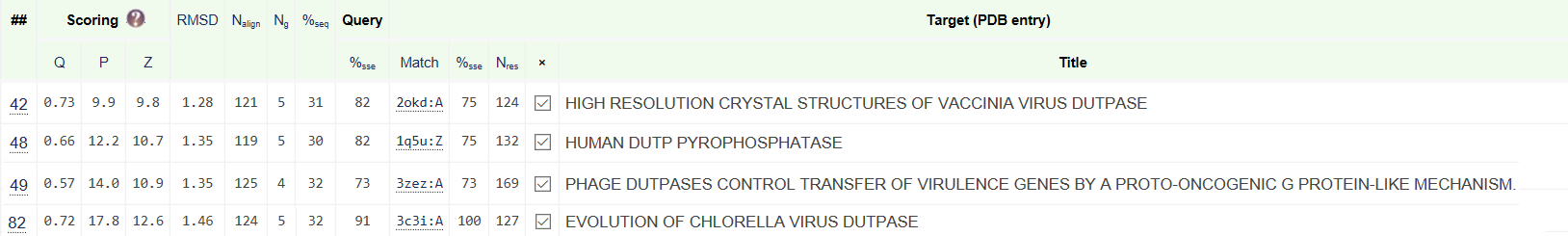 |
| Рис 1. Выбранные гомологи. |
Задание 2 |
| Далее с помощью PDBeFold было получено множественное выравнивание всех 5 структур. |
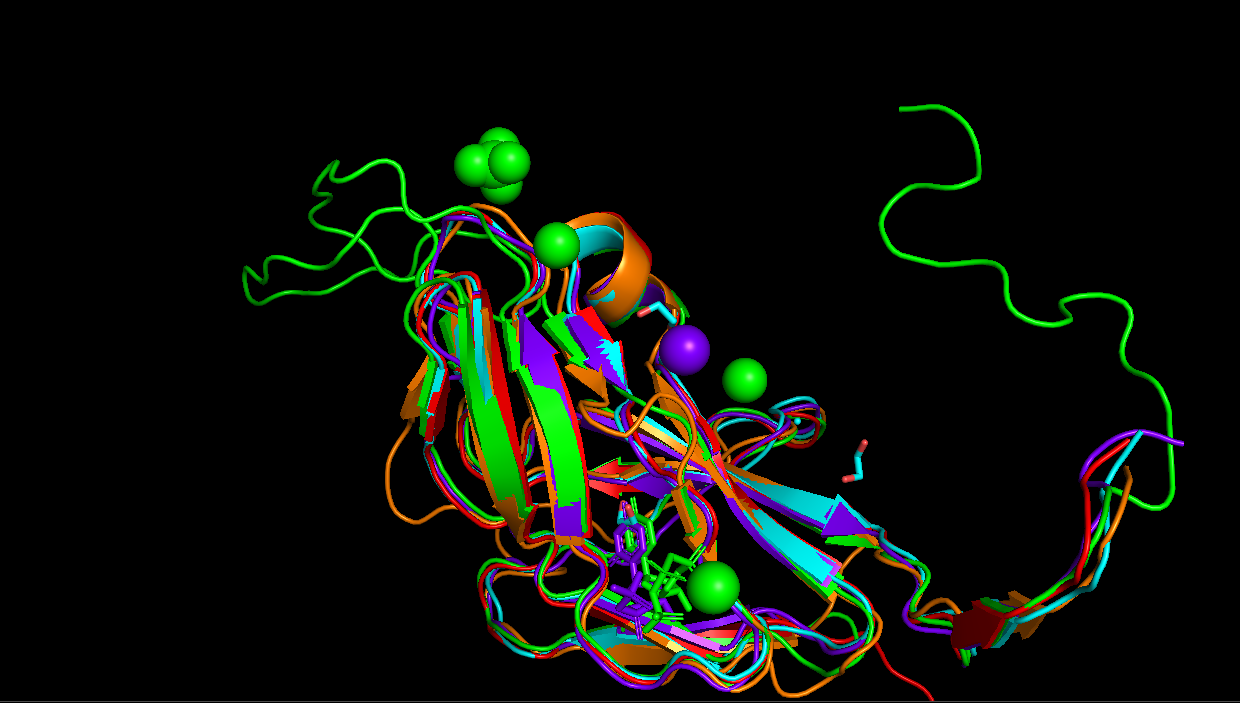 |
| Рис 2, Результаты структурного выравнивания (выравнивания последовательноcтей) предствалены в данном файле. |
| Все структуры хорошо совместились, включая образ 3ZEZ, если не учитвать петель (даеый белок гораздо длиннее остальных белков) |
| Затем было выполнено выравнивание при помощи Muscle (сверху) и споставленно структурному выравниванию. |
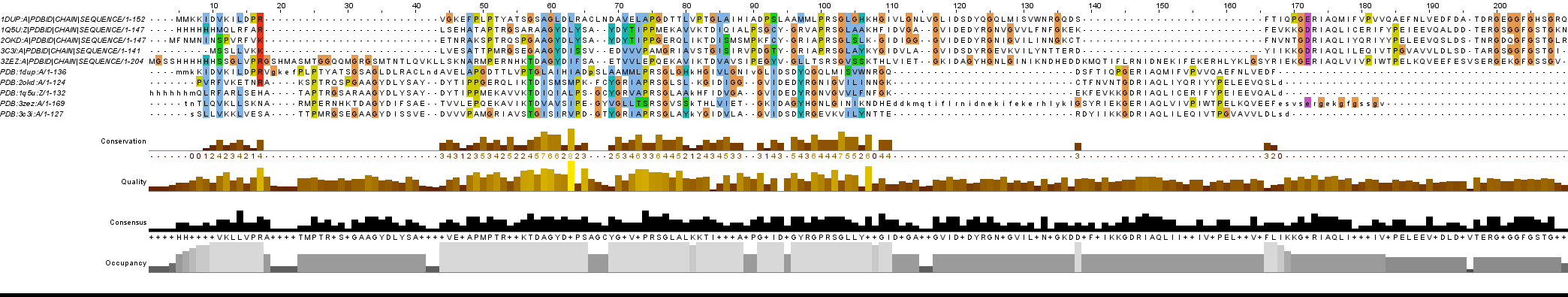 |
| Рис 3, результат выранивиний и их сравнение |
| Как видно на рисунке выше, существуют серьезные различия в выравнивании последовательностей по структуре от выравнивания тех же последовательностей, построенного программой |
| множественного выравнивания Muscle. В основном - это расположение гэпов, однако это очень сильно повлияло на то, как именно будут сопотставляться а.к. |
| Выравнивание PDBeFold по структурному совмещению, визуализированое с помощью Jalview (изображение в полном размере доступно по этой ссылке): |
| Для того, чтобы выяснить, какой метод точнее, была использована визуализация: |
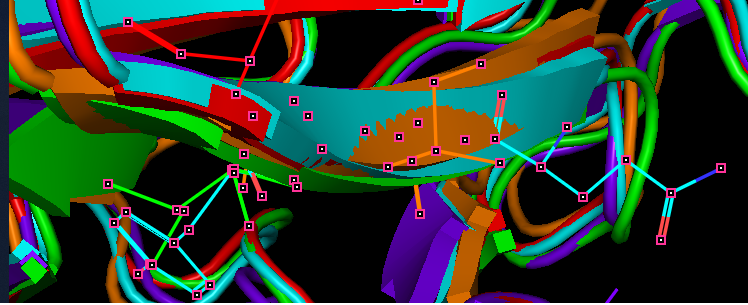 |
| Рис 4, визуализация |
| Так как выравнивания сильно различаются, то сравнивать по точечно не имеет смысла. Поэтому было выдвинуто предположение, что |
| удлинение белка скорее всего будет подразумевать более длиные петли (потеря консервативных остков нарушило бы функциональность белка) |
| Из этого следует, что гэпы выставленые в структрном выравнивании будут ближе к истине (т.к не будут разбивать консервативные участки, а петли |
| столь требовательны к постоянству а.к повледовательности). Поэтому был выбран участок, который в выранивании |
| программой разбит гэпом, а во вструктурном вырании является консервативным (нумерация из-за гэпов сильно съехала поэтому изначально предсказать более точное |
| соотвествие нумерации а.к было очень сложно). Оказалось, что данный участок, является бета листом, присутствующим во всех а.к (в том числе и в удлиненном |
| белке). Таким образом, метод структурного выравнивания сумел верно определить консервативные участки, в то время как muscle, аьсолютно не справился. |
| Оставшиеся различия никак не влияют на вывоД: выравнивание с помощью PDBeFold более достоверное. Оно намного лучше соответствует положению |
| аминокислотных остатков в белке. Программа множественного выравнивания |