Выравнивание последовательностей в JalView
Выравнивание последовательностей гомологичных белков
Выравнивание последовательностей - биоинформатический метод, основанный на размещении двух или более последовательностей мономеров ДНК, РНК или белков друг под другом таким образом, чтобы легко увидеть сходные участки в этих последовательностях. Сходство первичных структур двух молекул может отражать их функциональные, структурные или эволюционные взаимосвязи [1]. В ходе выполнения данной работы были выбраны шесть последовательностей гомологичных белков: два белка из домена Archaea (DNAK_METBU, DNAK_HALSA), два из домена Bacteria (DNAK_LACC3, DNAK_BORPD), два из домена Eukaryota (BIP2_MAIZE, MD37B_ARATH). С помощью программы JalView было постороено выравнивание этих последовательностей, для чего сначала из банка данных они были скачаны в fasta-формате, а затем импортированы в программу. На Рисунке 1 представлен результат выравнивания, где цветом выделены позиции, консервативные на 70% и более, то есть в данной позиции в одной из последовательностей одна аминокислота может отличаться от остальных.
 |
Рис 1. Выравнивание последовательностей гомологичных белков |
Можно заметить, что в данное выравнивание была добавлена дополнительная разметка - Markup. В этом поле отмечались литеры, каждая из которых означает следующее: C - позиции, консервативные (то есть в позиции один и тот же остаток) на 80% (то есть в позиции один остаток может быть другим) и более процентов, F - функционально консервативные позиции и G - позиции, где есть пропуски ("гэпы"). Функционально консервативные позиции - те, в которых аминокислотные остатки обладают сходными свойствами, в частности в 95 позиции в последовательности белка археи стоит аспартат, а в остальных - глутамат, они оба являются отрицательно заряженными; в 107 - находятся гидрофобные остатки аланина, изолейцина и валина; в 113 - гидрофильные остатки треонина и серина; в 114 - гидрофобные лейцин, валин и изолейцин. При смене в последовательностях гидрофобных аминокислот между собой, скорее всего, свойства белков в значительной мере не меняются, так как они обладают сходными функциями. В Таблице 1 представлена информация о консервативности белков, полученная с помощью программы infoalign из пакета EMBOSS, позволяющая рассчитать число и процент абсолютно консервативных позиций, в которых стоят одни и те же аминокислотные остатки, число и процента абсолютно функционально консервативных позиций, в которых во всех последовательностях стоит остаток из одной и той же группы (например, все гидрофобные/гидрофильные/заряженные), число и процент позиций с гэпами, число и процент позиций, консервативных на 70% и более.
| Таблица 1. Параметры консервативности. | ||||||||||
| Название последовательности | Длина | Длина выравнивания | Абсолютно консервативные позиции | % | Функционально консервативные позиции | % | Gaps | % | Консервативные на 70% | % |
| BIP2_MAIZE | 663 | 720 | 186 | 25.83 | 295 | 40.97 | 47 | 6.62 | 302 | 42.54 |
| DNAK_BORPD | 640 | 720 | 186 | 25.83 | 295 | 40.97 | 36 | 5.33 | 317 | 46.89 |
| DNAK_HALSA | 629 | 720 | 186 | 25.83 | 295 | 40.97 | 48 | 7.09 | 298 | 44.02 |
| DNAK_LACC3 | 624 | 720 | 186 | 25.83 | 295 | 40.97 | 54 | 7.96 | 315 | 46.46 |
| DNAK_METBU | 620 | 720 | 186 | 25.83 | 295 | 40.97 | 57 | 8.42 | 316 | 46.68 |
| MD37B_ARATH | 675 | 720 | 186 | 25.83 | 295 | 40.97 | 45 | 6.25 | 298 | 41.39 |
| Выравнивание | 720 | 186 | 25.83 | 295 | 40.97 | 99 | 13.75 | 263 | 36.53 | |
Ссылка на проект выравнивания.
Эволюция последовательности белка
Для выполнения данной работы был выбран участок последовательности белка α-L-фукозидазы (Alpha-L-fucosidase), характерного для организма Arthrobacter sp. A3 из рода Arthrobacter, начиная с 61 и заканчивая 161 аминокислотным остатком. Это белок является лизосомальным ферментом, участвующим в деградации различных групп естественных фукоглюкоконъюгатов [2]. С помощью программы msbar из пакета EMBOSS были проведены семь раундов мутаций, а затем полученные последовательности были выровнены в JalView. На Рисунке 2 представлен результат такого выравнивания, а ниже приведены комментарии к нему (буквами p* обозначены мутантные последовательности). В силу того, что программа может ошибаться, необходимо проверять, соответствует ли выравнивание эволюционному процессу. На Рисунке 3 представлен исправленный результат выравнивания с соответствующими комментариями.
Рис 2. Эволюция последовательности белка. Выравнивание |
Рис 3. Эволюция последовательности белка. Исправленное выравнивание |
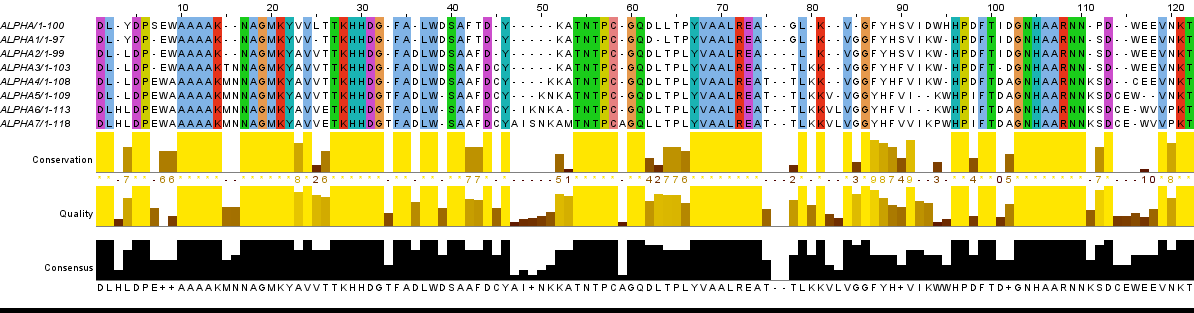 |
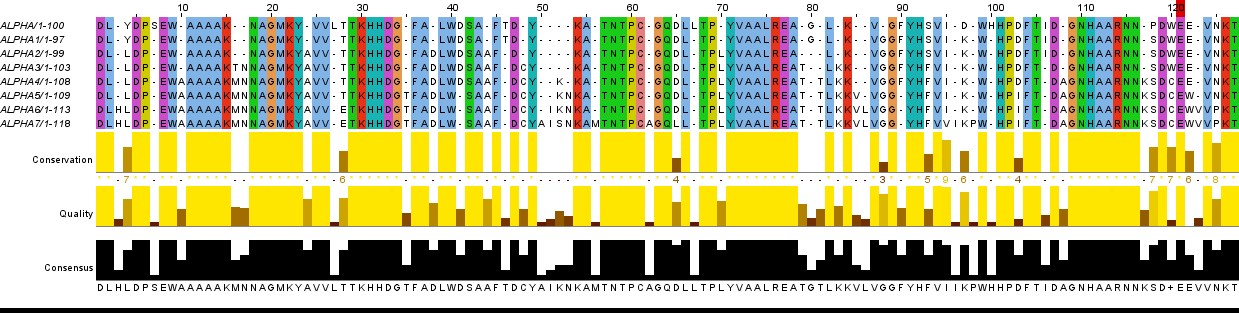 |
|
|
Ссылка на выравнивание. Ссылка на исправленное выравнивание. Ссылка на проект выравнивания.
Ссылка на скрипт, с помощью которого были проведены точечные мутации программой msbar.
Эволюция нуклеотидной последовательности
Для выполнения данной работы была взята нуклеотидная последовательность белка, транспортирующего тяжелые металлы (Heavy metal transporter), из протеома бактерии Arthrobacter sp.A3, который был изучен ранее. Информацию об этом белке можно найти в базе данных Uniprot, используя идентификатор CDS: ALV46833.1. Согласно представленным в ней данным размер белка составляет 75 аминокислотных остатков, а молекулярная масса - 7535 kDa; он является предсказанным (т.к. ему присвоен 4 номер), добавлен в базу данных Uniprot 16 марта 2016 года. Помимо этого, в анализируемом белке выделяется домен HMA с 4 по 72 аминокислотные остатки, а связывание металла предположительно осуществляется 14 и 17 остатками [3]. Нуклеотидная последовательность, с которой происходит транскрипция, а затем трансляция белка (Heavy metal transporter) располагается на комплементарной цепи геномной ДНК. Ниже представлены в fasta-формате нуклеотидная и аминокислотная последовательности рассматриваемого белка.
>ALV46833.1 heavy metal transporter [Arthrobacter sp. A3] MSHTITVNVSGMTCGHCVSSVTEELTGLKGVQDVAIDLNAGGISEVTITSSLTLDPAEIG EAVAEAGYVVVSNNA >NZ_CP013745.1:3660381-3660608 Arthrobacter sp. A3, complete genome, complementary chain TTATGCATTGTTGGAAACGACGACGTAGCCGGCCTCCGCGACGGCTTCGCCAATCTCCGC GGGATCAAGTGTCAGGGATGAGGTGATGGTGACTTCGGAAATGCCGCCGGCATTGAGGTC GATGGCGACGTCCTGGACGCCTTTGAGGCCCGTTAGTTCCTCCGTGACGGAGCTGACGCA GTGGCCGCAGGTCATGCCGGAAACGTTGACGGTGATGGTGTGGCTCAT |
С помощью программы msbar из пакета EMBOSS были проведены семь раундов мутаций в нуклеотидных последовательностях, а благодаря программе transeq из пакета EMBOSS была осуществлена трансляция данной полученной мутированной последовательности, а затем последовательности белков были выровнены в JalView. На Рисунке 4 представлен результат такого выравнивания, а ниже приведены комментарии к нему. Цветом выделены консервативные позиции, количество которых существенно меньше, чем можно наблюдать в предыдущем пункте работы. В силу того, что программа может ошибаться, необходимо проверять, соответствует ли выравнивание эволюционному процессу. На Рисунке 5 представлен исправленный результат выравнивания с соответствующими комментариями.
Рис 4. Эволюция нуклеотидной последовательности. Выравнивание |
Рис 5. Эволюция нуклеотидной последовательности. Исправленное выравнивание |
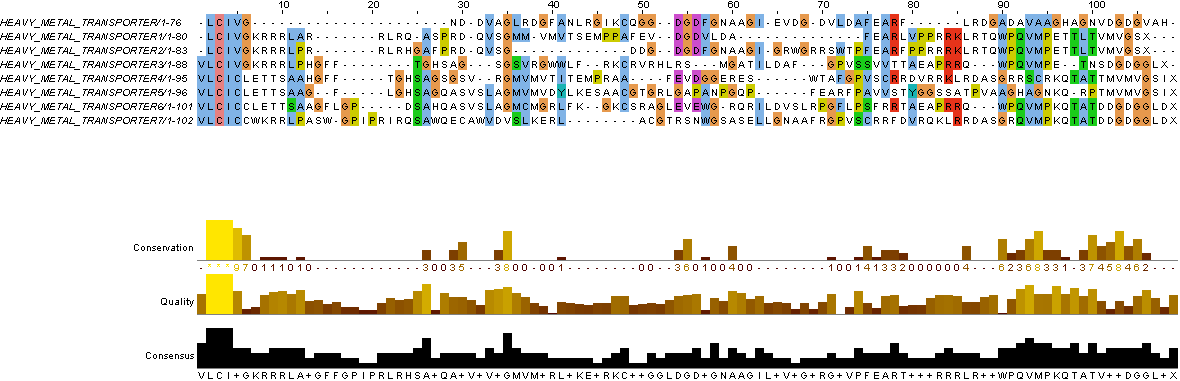 |
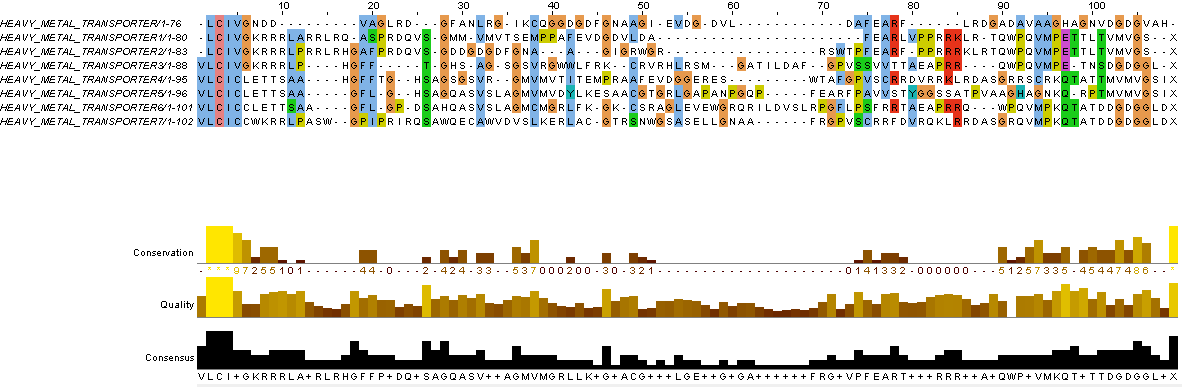 |
|
|
Ссылка на выравнивание. Ссылка на исправленное выравнивание. Ссылка на проект выравнивания. Ссылка на скрипт, с помощью которого были проведены точечные мутации программой msbar.
Выводы
Если сравнить результат мутации аминокислотной последовательности и нуклеотидной, то можно заметить, что во втором случае выравниванию, выполненному автоматически и вручную, верить, скорее всего, нельзя, и оно не является оптимальным, так как в нём присутствует большое количество гэпов, мало консервативных позиции и не представляется возможным выделить блоки. Из этого следует, что точечные мутации в нуклеотидной последовательности обуславливают более фатальные функциональные изменения в белке, чем мутации в аминокислотной последовательности. Можно предположить, что такая разница объясняется сдвигом рамки считывания, который происходит при каждой точечной мутации в нуклеотидной последовательности, как-то: делеция или вставка нуклеотидов. Из-за произошедшего изменения в нуклеотидной последовательности кардинальным образом изменилась последовательность белка, а консервативные позиции почти все исчезли, поэтому приведенное выше выравнивание является бессмысленным.
Take home messages
"Эволюционирует нуклеотидная последовательность генома." Биологическая эволюция - естественный процесс развития живой природы, сопровождающийся изменением генетического состава популяций, формированием адаптаций, видообразованием и вымиранием видов, преобразованием экосистем и биосферы в целом [4]. Зачастую эволюционный процесс протекает на уровне организма, то есть в популяции происходит естественный отбор, по результатам которого сохраняются те или иные мутации, являющиеся выгодными для существования в данных условиях обитания и способствующие увеличению числа потомков с данными признаками в популяции, благодаря чему и происходит закрепление мутировавшей аллели. Таким образом, эволюционирует белок, так как именно он находится под действием отбора. Например, белки с одинаковой последовательностью, могут иметь разную конформацию, обуславливающую разность их функций. Так, существуют прионы — особый класс инфекционных агентов, представленных белками с аномальной третичной структурой и не содержащих нуклеиновых кислот. Отличительной их особенностью является то, что в организме существует нормальная форма белка со свойственной ему третичной структурой, но в организме могут возникать изоформы этой белковой пследовательности, обусловленные отличной от нормальной третичной структурой и я вляющиеся патогенами. Этот пример подтверждает, что эволюционирует белок [5].
"Гомологичность последовательностей белков и их а.к.о. можно предсказать по высокому сходству фрагментов в блоках выравнивания." Как правило, происходит именно так, однако существует такое биологическое явление, как аналогия - внешнее сходство организмов разных систематических групп, а также органов или их частей, происходящих из различных исходных зачатков и имеющих неодинаковое строение. Аналогия обусловлена общностью образа жизни или функции [6]. И в таком случае в блоках могут располагаться довольно большое количество сходных фрагментов, так как белки эволюционно выполняющие одинаковые функции обладают сходным аминокислотным составом, зачастую за счет большого количества функционально консервативных позиций в последовательности. Однако они не произошли от одного предка и гомологичными не являются, но в блоках наблюдается сходство аминокислотного состава. Например, у млекопитающих и дрожжей белки синаптонемного комплекса (синаптонемный комплекс - структура, формирующаяся во время мейоза)имеют разные аминокислотные последовательности, но их вторичная и третичная структуры одинаковы. Так, белок «застежки-молнии» SCP1 у млекопитающих и негомологичный ему белок Zip1 у дрожжей построены по единому плану [7].
Источники:
| [1] Выравнивание последовательностей - wikipedia.org [2] Фукозидазы - wikipedia.org [3] Uniprot [4] Эволюция - wikipedia.org [5] Прионы [6] Аналогия - wikipedia.org [7] Синаптонемный комплекс |