Поиск полиморфизмов у человека
Дано
-
Чтения экзома, кртирующиеся на участок хромосомы человека - chr2.fastq
-
Хромосомы человеческого генома (сборка версии hg19) - chr2.fasta
Часть I: подготовка чтений
Анализ качества чтений
С помощью программы FastQC был проведён контроль качества чтений. Команда:
fastqc chr2.fastq |
Получив на вход файл в формате .fastq (данный формат характерен для файлов с данными после NGS), программа выдала архив (.zip) с отчетом по работе в виде текстового файла и рисунков, в котором помимо прочей информации находился файл chr2_fastqc.html, содержащий визуализацию качества чтений.
Очистка чтений
Используя программу Trimmomatic, производилась очистка чтений. Команда:
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr2.fastq chr2_clean.fastq TRAILING:20 MINLEN:50 |
Здесь, параметр TRAILING отвечает за удаление нуклеотидов с конца каждого чтения, если их quality score меньше, чем 20, а параметр MINLEN оставляет риды длиной 50 и более, а риды с меньшей длиной удаляются. В результате число прочтений уменьшилось с 10410 до 10191, то есть на 2,1%. Далее, с помощью FastQC был вновь проведен анализ качества чтений и получен архив с файлом - chr2_clean_fastqc.html. Команда:
fastqc chr2_clean.fastq |
В Таблице 1 приведены различия по чтениям до и после очистки.
Таблица 1. Характеристика ридов до и после очистки. | ||
Категория |
До очистки |
После очистки |
Число ридов |
10410 |
10191 |
Длина ридов |
40-100 |
50-100 |
%GC |
42 |
42 |
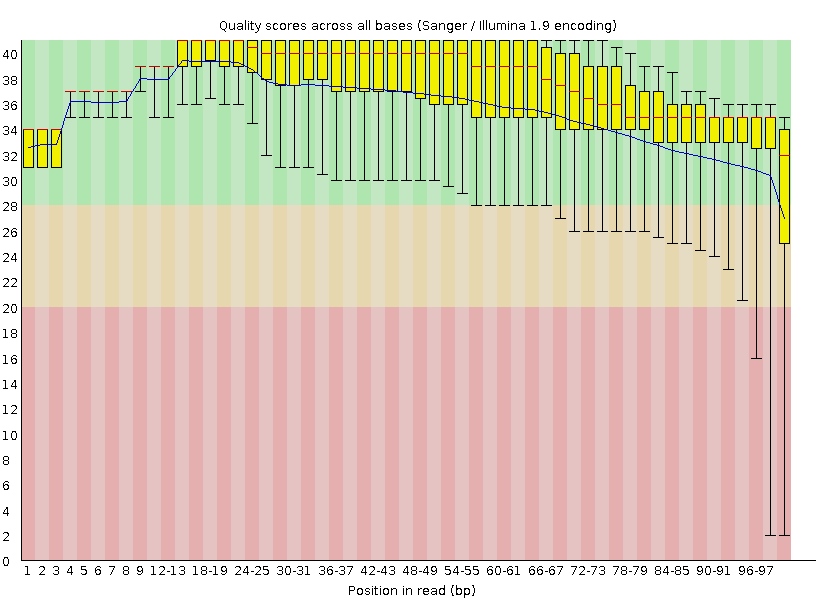
На Рис. 1-2 представлена информация, полученная с помощью программы FastQC, а именно - характеристика качества каждого нуклеотида в ридах. По абсциссе расположены позиции в ридах, а по ординате - Quality score, причем данные представлены в виде диаграмм boxplot, где границами желтых столбцов служат первый и третий квартили, красная линия - медиана, синяя линия - среднее значение, а отходящие от столбцов "усы" демонстрируют размах между 10-м и 90-м процентилями. Помимо этого само поле графика покрашено в три цвета: нахождение диаграмм в зеленой плотности говорит о том, что качество данной позиции хорошее и она достоверна (выше 28), в оранжевой - среднее (от 20 до 28), в красной - плохое, таким последовательностям верить нельзя (от 0 до 20).
|
На Рис. 1 можно учидеть, что в 14 позициях "усы" диаграмм попадают в оранжевую область, а в трех уходят в красную, такой результат говорит о падении качества чтения последних нуклеотидов. Однако, в целом, большая часть позиций обладает высоким качеством прочтения. |
Рис. 1. Характеристика качества последовательностей ридов до очистки. | |
|
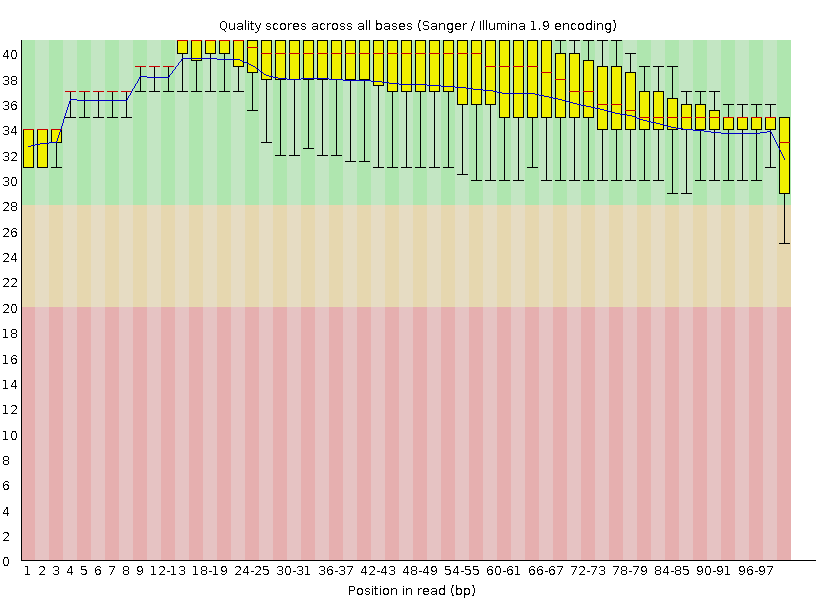
Как и следовало ожидать, после очистки исчезли концевые нуклеотиды, обладающие quality score < 20, а следовательно, на графике это отразилось "укорачиванием усов" нуклеотидов, особенно значительно это для последних позиций. Теперь лишь в последней позиции диаграмма заходит в оранжевую область, а такую совокупность ридов можно достоверно использовать дальше. |
Рис. 2. Характеристика качества последовательностей ридов после очистки. |
Помимо этого, как уже отмечалось выше, программа FastQC выдает различные графики, ниже на Рис. 3-4 представлена характеристика распределения quality score среди чтений.
|
|
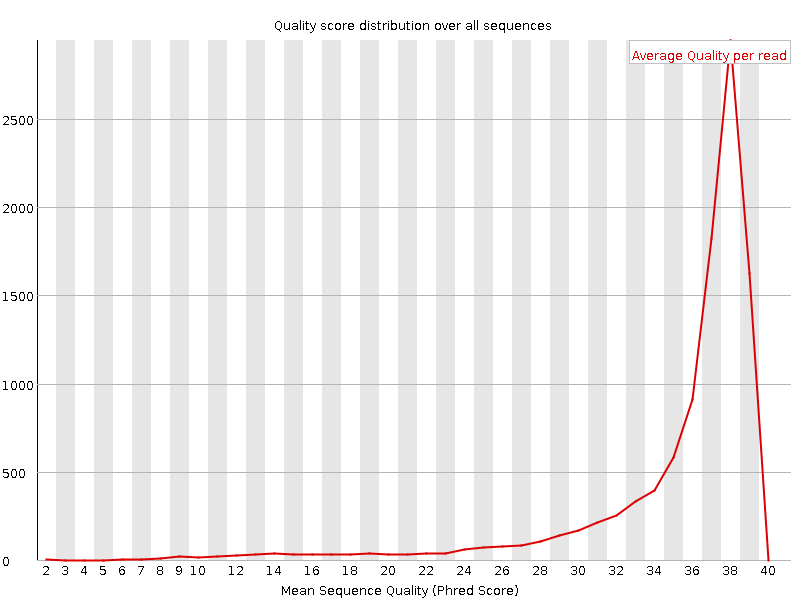
Рис. 3. Распределение среднего качества до очистки. |
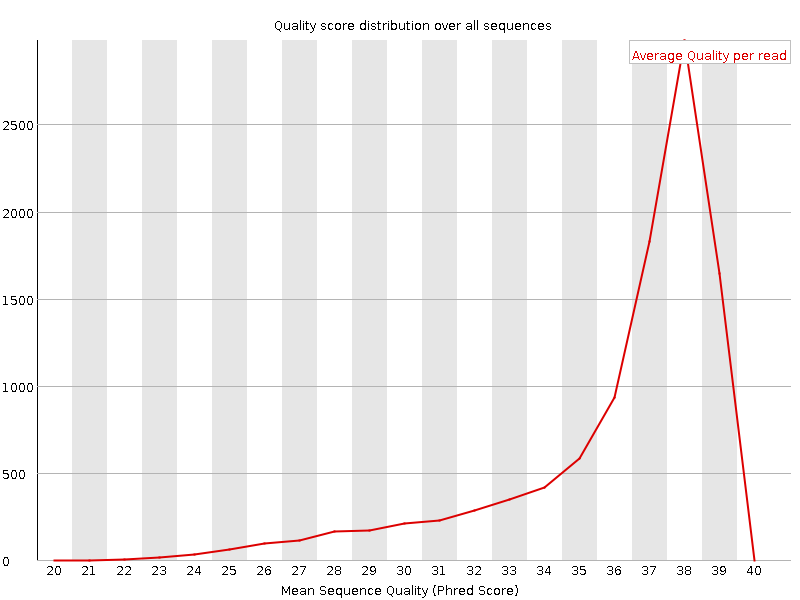
Рис. 4. Распределение среднего качества после очистки. |
Как видно из Рис. 3, до очистки среди всех ридов присутствуют чтения с качеством ниже 20, однако большая часть обладает качеством выше 30, что является очень хорошим показателем. |
После очистки можно увидеть, что среди всех ридов отсутствуют рида качеством ниже 20, а превалирующая часть чтений всё так же обладает средним качеством выше 30. |
Часть II: картирование чтений
Картирование чтений
С помощью программы Hisat2 было осуществлено картирование чтений. Сначала была проиндексирована референсная последовательность с помощью команды:
hisat2-build chr2.fasta chr2 |
В результате чего были получены 8 файлов в формате .ht2. Затем построено выравнивание очищенных чтений и референса в формате .sam с помощью команды, где параметр --no-softclip запрещает подрезать чтения, а --no-spliced-alignment требует проводить картирование без разрывов:
hisat2 -x chr2 -U chr2_clean.fastq --no-spliced-alignment --no-softclip > align_clean.sam |
В результате выдача сохранена в файл align_clean.sam.
Анализ выравнивания
С помощью команды, приведенной ниже, выравнивание из формата .sam было переведено в бинарный формат .bam:
samtools view align_clean.sam -bo align_clean.bam |
Для этого использовалась команда view из пакета samtools. Затем выравнивание чтений с референсом было отсортировано по координате в референсе начала чтения с помощью следующей команды:
samtools sort align_clean.bam -T 1.txt -o chr2_sort.bam |
И далее отсортированный файл chr2_sort.bam был проиндексирован командой:
samtools index chr2_sort.bam |
После сего получен файл chr2_sort.bam.bai. Затем выяснялось, сколько чтений откартировано на геном, эта информация выдавалась сразу после работы программы Hisat2:
10191 reads; of these:
10191 (100.00%) were unpaired; of these:
48 (0.47%) aligned 0 times
10139 (99.49%) aligned exactly 1 time
4 (0.04%) aligned >1 times
99.53% overall alignment rate
Из которой видно, что 48 чтений не было совсем откартировано, 10139 откартировалось ровно 1 раз, а 4 - больше, чем один раз. В то же время эту информацию можно получить с помощью команды:
samtools idxstats chr2_sort.bam > out.txt |
В результате в файле out.txt была записана следующая информация: длина референсной последовательности - 243199373 и число откартированных чтений, однако никаких пояснений не даётся.
Дополнительно
Для выполнения данного задания с помощью команды, приведенной ниже, было вычислено покрытие для каждого из нуклеотидов:
samtools depth chr2_sort.bam > depth.txt |
Затем в полученном файле depth.txt был выбран нуклеотид с покрытием 206 в позиции 234189668. С помощью GenomeBrowser (версия генома hg19) определены координаты экзона, содержащего выбранный нуклеотид: 234160217-234204320. После этого была перезапущена команда samtools depth с указанием координат экзона:
samtools depth -r chr2:234160217-234204320 chr2_sort.bam > depth_ex.txt |
И получен файл depth_ex.txt, в котором обозначалось вычисленное покрытие для нуклеотидов из экзона, а с помощью программы Excel вычислялось среднее покрытие чтениями, оказавшееся равным 31,1568.
Часть III: Анализ SNP
Поиск SNP и инделей
С помощью команды:
samtools mpileup -uf chr2.fasta chr2_sort.bam > snp.bcf |
Был создан файл с полиморфизмами в формате .bcf. Затем командой:
bcftools call -cv snp.bcf > snp.vcf |
Получен файл со списком отличий между референсом и чтениями в формате .vcf. В файле snp.vcf по результатам работы программы bcftools call было найдено 79 полиморфизмов, из которых 7 инделей и 72 замены. В Таблице 2 собрана информация о 3 полиморфизмах.
Таблица 2. Описание полиморфизмов. | |||||
Координата |
Тип полиморфизма |
Референс |
Чтения |
Глубина покрытия |
Качество чтений |
234181593 |
Замена |
G |
C |
28 |
134.008 |
234202274 |
Вставка |
TCC |
TCCC |
1 |
3.80767 |
238426328 |
Делеция |
gtggatggatggatggatggatggatggatggat |
gtggatggatggatggatggatggatggat |
1 |
22.4955 |
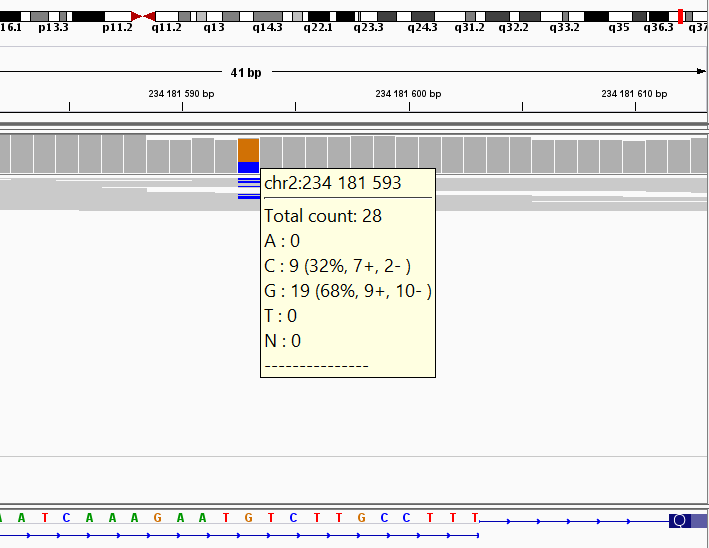
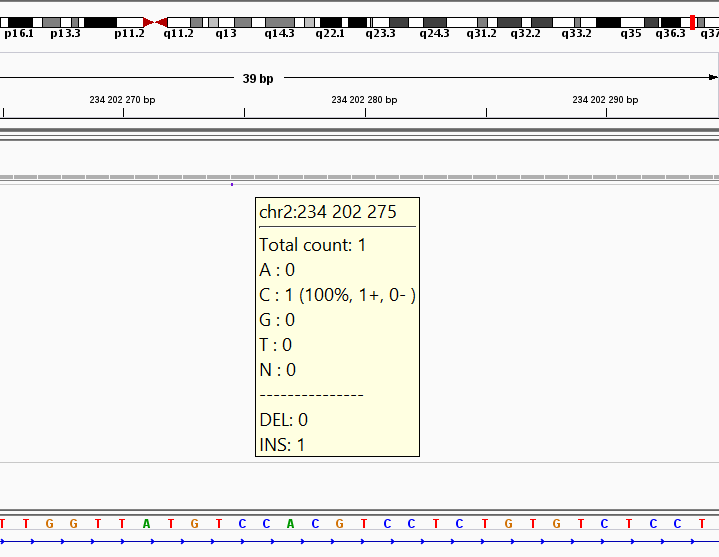
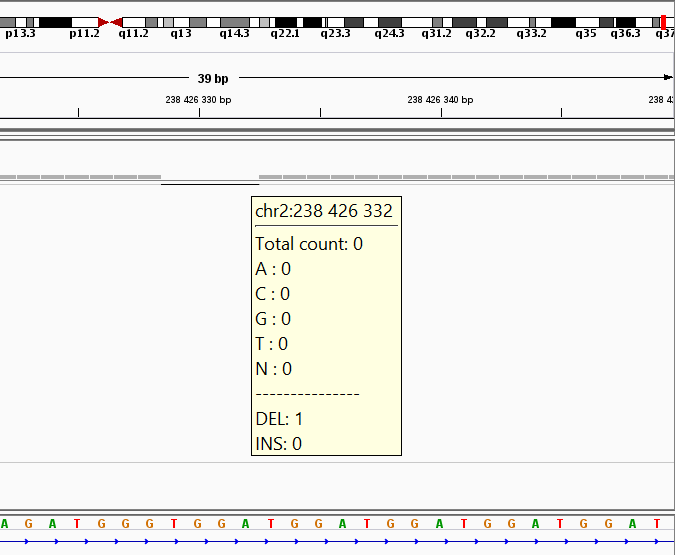
С помощью программы IGV - Integrative Genomics Viewer, были визуализированы полиморфизмы, приведённые в Таблице 2. На вход программа получает отсортированный файл с выравниванием в формате .bam. Рис. 5-7 демонстрируют результат работы этой программы, для каждого полиморфизма программа выдает краткую информацию. Верхней линией на рисунках показана картированная хромосома, что позволяет довольно легко найти нужный участок.
|
|
|
Рис. 5. Замена 234181593. |
Рис. 6. Вставка 234202274. |
Рис. 7. Делеция 238426328. |
Программа IGV выдает информацию о том, в каком числе ридов встречается тот или иной нуклеотид, например, в данном случае видно, что цитозин встречается в 9 из 28 чтений, а гуанин в 19 из 28, в референсной последовательности гуанин. |
В данном примере произошедшая вставка описана в пояснениях и показана синей точкой. |
Здесь показана делеция в пояснениях и в виде пропуска нуклеотидов в покрытии. |
Аннотация SNP
Программа annovar позволяет аннотировать snp, однако сначала необходимо перевести файл в формат, с которым способна работать данная программа. Сперва были удалены индели, следовательно в файле осталось только 72 snp, затем с помощью приведенной ниже команды файл snp.vcf был переведён в файл snp.avinput.
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp.vcf -outfile snp.avinput |
Затем проводилось аннотирование полиморфизмов по следующим базам данных:
-
refgene - gene-based annotation
-
dbsnp - filter-based annotation
-
1000 genomes - filter-based annotation
-
Gwas - region-based annotation
-
Clinvar - filter-based annotation
1. dbSNP
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs_find.snp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/ |
По результатам работы программы annovar были получены три файла:
-
rs_find.snp.log - описание работы программы
-
rs_find.snp.hg19_snp138_dropped - список аннотированных в базе данных snp (имеющих rs)
-
rs_find.snp.hg19_snp138_filtered - список не аннотированных в базе данных snp (не имеющих rs)
снено, что 69 полиморфизмов имеют rs, а 3 не имеют. По этой ссылке Вы можете ознакомиться с примером аннотации полиморфизма в базе данных. Здесь описывается, из каких источников была получена информация о наличии полиморфизмов, характерность встречаемости данных полиморфизмов в популяциях и другая полезная информация. Неаннотированные полиморфизмы характеризуются низким качеством чтения (< 10).
2. RefGene
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -out rs.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ |
По результатам работы программы annovar были получены три файла:
-
rs.refgene.log - описание работы программы
-
rs.refgene.exonic_variant_function - описание полиморфизмов, попавших в экзоны (часть информации из файла rs.refgene.variant_function)
-
rs.refgene.variant_function - информация о расположении полиморфизмов относительно генов (в нетранслируемых областях, в интронах, в экзонах и др)
В Таблице 3 приведена характеристика полиморфизмов по базе данных refgene.
Таблица 3. Расположение snp. | ||
Зона |
Описание |
Число snp |
UTR3 |
snp в 3'нетранслируемой области |
5 |
intronic |
snp в интронах |
57 |
UTR5 |
snp в 5'нетранслируемой области |
2 |
exonic |
snp в экзонах |
6 |
ncRNA_exonic |
snp в транскрибируемой РНК, не имеющей аннотированного кодирующего участка |
1 |
Из данной информации следует, что наибольшее количество полиморфизмов приходится на интроны. Примерно одинаковое число полиморфизмов приходится на экзоны и 3'нетранслируемую область. Помимо этого, присутствует информация, является ли данный полиморфизм гетерозиготным (het - 30) или гомозиготным (hom - 42). Информация о полиморфизмах, попавших в экзоны представлена в Таблице 4.
Таблица 4. Характеристика snp, попавших в экзоны. | ||||||||
Ген |
Координата |
Референс |
Чтение |
Синонимичность замены |
Тип замены |
Глубина |
Качество |
Аминокислотная замена |
ATG16L1 |
234183368 |
A |
G |
Несинонимичная |
Гетерозиготная |
41 |
225.009 |
T184A |
MLPH |
238427194 |
T |
C |
Несинонимичная |
Гетерозиготная |
22 |
192.009 |
L153P |
MLPH |
238427251 |
G |
A |
Несинонимичная |
Гетерозиготная |
24 |
131.008 |
G172D |
MLPH |
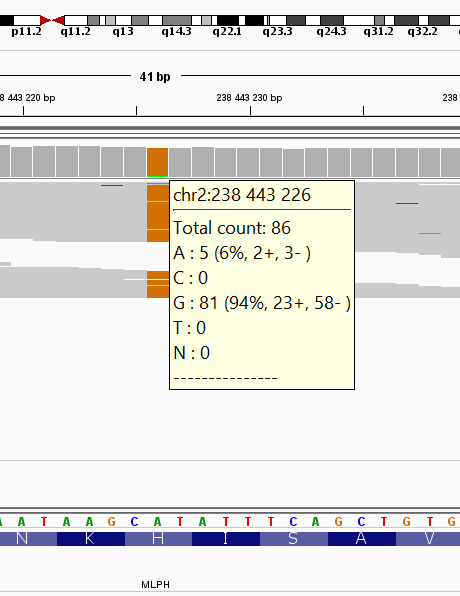
238443226 |
A |
G |
Несинонимичная |
Гомозиготная |
86 |
221.999 |
H347R |
MLPH |
238449007 |
T |
C |
Несинонимичная |
Гомозиготная |
17 |
224.127 |
V374A |
MLPH |
238449107 |
A |
G |
Синонимичная |
Гомозиготная |
13 |
219.003 |
E407E |
Обозначенные в Таблице 4 гены соответствуют: MLPH (NM_024101) - ген, кодирующий белок, участвующий в связывании пигментных органелл, называемых меланосомами, с актиновым цитоскелетом в мелоноцитах, необходимый для видимой пигментации в волосах и коже [1]; ATG16L1 (NM_017974) - ген, кодирующий белок, являющийся частью белкового комплекса, необходимого для аутофагии - основного процесса, благодаря которому внутриклеточные компоненты деградируют с помощью лизосом [2].
3. 1000 genomes
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.1000g -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/ |
По результатам работы программы annovar были получены три файла:
-
rs.1000g.log - описание работы программы
-
rs.1000g.hg19_ALL.sites.2014_10_dropped - информация о частоте встречаемости конкретных snp
-
rs.1000g.hg19_ALL.sites.2014_10_filtered - информация о snp, для которых не известна частота встречаемости
В результате работы программы для трех полиморфизмов не было найдено информации о частоте встречаемости, для остальных 69 она указана, причем наибольшая частота встречаемости равна 0,994209, а минимальная - 0,00738818. Информация по частоте встречаемости полиморфизмов, попавших в экзоны представлена в Таблице 5 (так как нас интересуют именно попавшие в экзоны полиморфизмы).
Таблица 5. Характеристика snp, попавших в экзоны. | ||||||||
Ген |
Координата |
Референс |
Чтение |
Тип замены |
Аминокислотная замена |
Частота встречаемости |
ATG16L1 |
234183368 |
A |
G |
Гетерозиготная |
T184A |
0.395966 |
MLPH |
238427194 |
T |
C |
Гетерозиготная |
L153P |
0.178714 |
MLPH |
238427251 |
G |
A |
Гетерозиготная |
G172D |
0.178714 |
MLPH |
238443226 |
A |
G |
Гомозиготная |
H347R |
0.371406 |
MLPH |
238449007 |
T |
C |
Гомозиготная |
V374A |
0.505591 |
MLPH |
238449107 |
A |
G |
Гомозиготная |
E407E |
0.505591 |
Как видно из Таблицы 5, частота встречаемости данных полиморфизмов довольно высока. Также с помощью программы Excel были подсчитаны среднее значение и медиана частот встречаемости полиморфизмов, они оказались равными 0,372 и 0,375 соответственно. Можно заметить, что в данном случае медиана и среднее значение оказались почти одинаковыми, из чего следует, что распределение частот встречаемости полиморфизмоф близко к нормальному.
4. Gwas
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out rs.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/ |
По результатам работы программы annovar были получены два файла:
-
rs.gwas.log - описание работы программы
-
rs.gwas.hg19_gwasCatalog - список snp, имеющих клиническое значение
В полученном файле содержалась информация по 4 из 72 полиморфизмам, значит, что лишь 4 могут иметь своё фенотипическое проявление. В Таблице 6 собрана информация по этим snp.
Таблица 6. Характеристика snp. | |||||||
Фенотипическое проявление | Координата | Референс | Чтение | Тип замены | Глубина | Качество | Расположение snp |
Рост | 55516323 | T | G | Гетерозиготная | 112 | 225.009 | UTR3 |
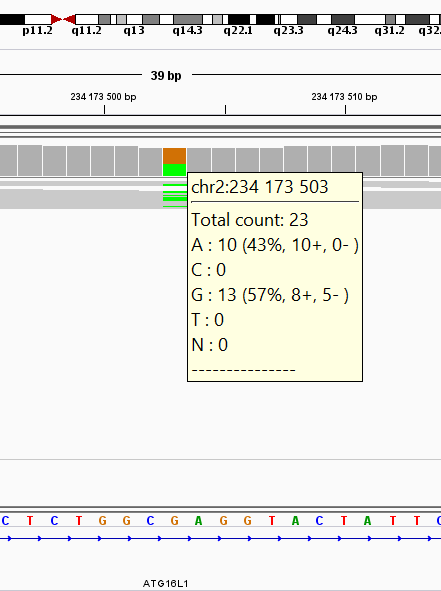
Болезнь Крона | 234173503 | G | A | Гетерозиготная | 23 | 113.008 | Интрон |
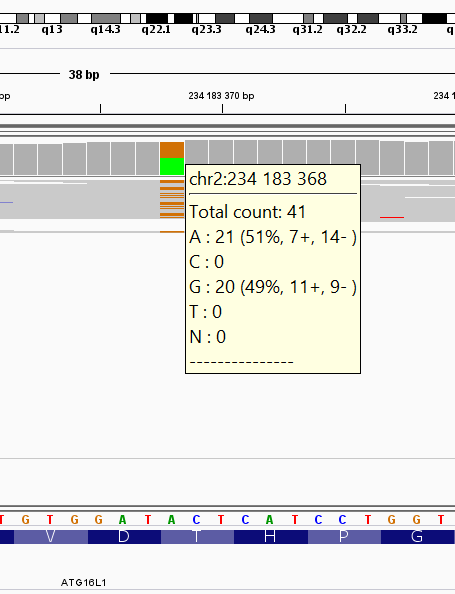
Болезнь Крона | 234183368 | A | G | Гетерозиготная | 41 | 225.009 | Экзон |
Рак простаты | 238443226 | A | G | Гомозиготная | 86 | 221.999 | Экзон |
Замечание: болезнь Крона (гранулематозный энтерит) - тяжёлое хроническое иммуноопосредованное гранулематозное воспалительное заболевание желудочно-кишечного тракта, которое может поражать все его отделы, начиная от полости рта и заканчивая прямой кишкой, с преимущественным поражением терминального отрезка подвздошной кишки и илеоколитом в 50 % случаев [3].
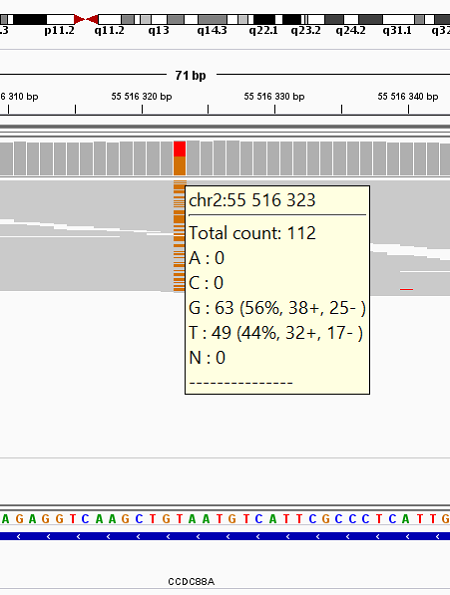
С помощью программы IGV приведенные в Таблице 6 полиморфизмы были визуализованы, так как, возможно, являются клинически значимыми и интересно узнать их расположение. На Рис. 8-11 показаны результаты такой работы. Довольно интересно, что в программе можно узнать, в какую зону входит полиморфизм, так, для первого полиморфизма видим, что под аминокислотной последовательностью нет аминокислотной последовательности, что согласуется с информацией, которая раньше была получена в базе данных Gwas: из неё следует, что данный snp входит в 3'нетранслируемую область, что отражено толстой синий полосой под нуклеотидной последовательностью, однако, если бы полиморфизм входил в состав интрона, полоса была бы тонкой, как это представлено на Рис. 9. Если полиморфизм входит в экзон, то под нуклеотидной последовательностью идёт аминокислотная, как на Рис. 10-11. Помимо этого, в отличие от первых трех полиморфизмов, у которых встречаемость разных нуклеотидов в чтениях составляет примерно 50 на 50, в последнем полиморфизме замена аденина на гуанин наблюдается в 81 из 86 чтениях, что является довольно значим результатом и согласуется с тем, что в базе данных Clinvar было найдено заболевание именно по этому полиморфизму, которое ассоциировано с данной мутацией.
|
|
|
|
Рис. 8. Фенотипическое проявление - рост. |
Рис. 9. Фенотипическое проявление - болезнь Крона (234173503). |
Рис. 10. Фенотипическое проявление - болезнь Крона (234183368). |
Рис. 11. Фенотипическое проявление - рак простаты. |
5. Clinvar
Команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.clinvar -dbtype clinvar_20150629 -buildver hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ |
По результатам работы программы annovar были получены три файла:
-
rs.clinvar.log - описание работы программы
-
rs.clinvar.hg19_clinvar_20150629_dropped - список snp, аннотированных в Clinvar
- r
s.clinvar.hg19_clinvar_20150629_filtered - список snp, не аннотированных в Clinvar
Лишь один полиморфизм является аннотированным в базе данных Clinvar: в позиции 234183368 замена T184A асооциирована с воспалительным заболеванием кишечника, этот результат согласуется с возможным фенотипическим проявлением болезни Крона, которое было аннотировано в базе данных Gwas. Остальные 71 замены не аннотированы в Clinvar. Таким образом, база данных Gwas дала более полный список возможных проблем, вызываемых присутствующими полиморфизмами. Была составлена сводная таблица, в которую вошли все snp и их характеристики по использованным для аннотации базам данных. Цветами выделены наиболее интересные и вероятные заболевания и проблемы, обусловленные данными полиморфизмами. Таблица доступна по этой ссылке snp.xlsx.
Распределение чтений
С помощью программы IGV было так же проведен анализ распределения чтений относительно экзонов, на Рис. 12 можно познакомиться с результатом этого анализа.
|
Рис. 12. Распределение чтений. |

Как видно на Рис. 12 чтения распределяются внутри экзонов и немного вокруг них, на далеком расстоянии от экзонов их нет (экзоны обозначены толстыми синими полосами, при увеличении изображения показывается аминокислотная последовательность).
Таблица 7. Команды использованные для выполнения данного практикума. | ||||||||
Команда |
Пояснение |
|||||||
fastqc chr2.fastq |
Проводит контроль качества чтений |
|||||||
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr2.fastq chr2_clean.fastq TRAILING:20 MINLEN:50 |
Очищает чтения |
|||||||
fastqc chr2_clean.fastq |
Проводит контроль качества очищенных чтений |
|||||||
export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 |
Доступ к файлам для запуска программы Hisat2 |
|||||||
hisat2-build chr2.fasta chr2 |
Индексирование референсной последовательности, выдает файлы в формате .ht2 |
|||||||
hisat2 -x chr2 -U chr2_clean.fastq --no-spliced-alignment --no-softclip > align_clean.sam |
строит выравнивание очищенных чтений и референса в формате .sam. Параметр --no-softclip запрещает подрезать чтения, а --no-spliced-alignment требует проводить картирование без разрывов. |
|||||||
samtools view align_clean.sam -bo align_clean.bam |
Переводит выравнивание в бинарный формат |
|||||||
samtools sort align_clean.bam -T 1.txt -o chr2_sort.bam |
Сортирует выравнивание чтений с референсом в формате .bam по координате в референсе начала чтения, в файл 1.txt записана выдача stdout после работы программы |
|||||||
samtools index chr2_sort.bam |
Индексирует сортированный файл |
|||||||
samtools idxstats chr2_sort.bam > out.txt |
Выводит информацию об откартированных чтениях на геном |
|||||||
samtools depth chr2_sort.bam > depth.txt |
Вычисляет покрытие каждого из нуклеотидов, и выводит эту информацию в файл depth.txt |
|||||||
samtools depth -r chr2:238395053-238463961 chr2_sort.bam > depth_ex.txt |
Вычисляет покрытие для каждого нуклеотида из указанной области |
|||||||
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp.vcf -outfile snp.avinput |
Переводит файл snp.vcf в формат, с которым может работать программа annovar |
|||||||
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs_find.snp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/ |
Позволяет узнать, какие snp имеют rs |
|||||||
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -out rs.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/ |
Поиск по Gwas |
|||||||
erl /nfs/srv/databases/annovar/annotate_variation.pl -out rs.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ |
Поиск по RefGene |
|||||||
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.clinvar -dbtype clinvar_20150629 -buildver hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ |
Поиск по Clinvar |
|||||||
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.1000g -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/ |
Поиск по 1000 genome |
|||||||
Источники:
| [1]Melanophilin [2]ATG16L1 [3]Болезнь Крона |