Учебный сайт Полины Байкузиной | |||
| Главная | Семестры | О себе | Ссылки |
Ресеквенирование. Поиск полиморфизмов у человекаЗадача: найти и описать полиморфизмы у пациента. Дано:
Часть I. Задание 1. Анализ качества чтений. Требовалось сделать контроль качества чтений с помощью программы FastQC. Выполненная команда представлена в табл.1. В результате работы программы был получен архив (chr3_fastqc.zip), который содержит отчет о программе в виде html файла (chr3_fastqc.html"). На рис.1. представлены изображение результатов работы программы FastQC, на котором показано качество определения нуклеотида в каждой позиции рида. 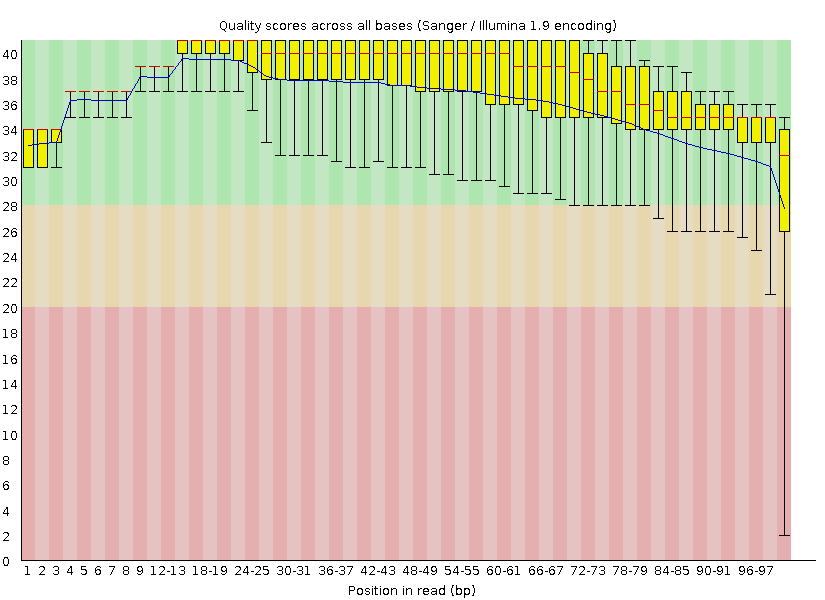
Элементы:
Поле графика разделено на 3 области (зеленый, желтый, красный), соответствующие качеству чтений (зеленый цвет - хорошее качество, красный - плохое). Как видно из изображения конец ридов прочитан хуже, чем основная часть (примерно с 84 позиции "усы" находятся в желтой области, в последней позиции выходят в красную область. На рис.2 представлена основная информация о чтениях. Как видно из таблицы, количество чтений до чистки 20932. Длина чтений 30-100. 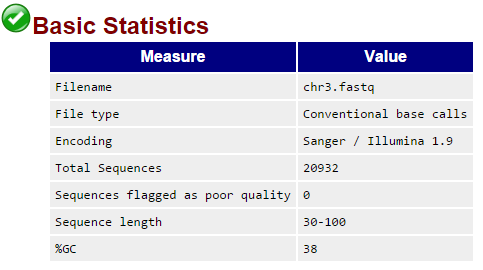
Задание 2. Очистка чтений. Для очистки чтений использовалась программа Trimmomatic. Нужно было отрезать с конца каждого чтения нуклеотиды с качеством ниже 20 и оставить только чтения длиной не меньше 50 нуклеотидов. Команда приведена в табл.1 и на рис.3. 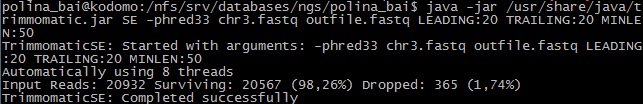
Как видно из рисунка после чистки осталось 20567 чтений; 365 чтений было удалено. Затем я снова запустила программу FastQC для того, чтобы проанализировать качество (команда: fastqc outfile.fastq; результат: html). На рис.4 изображено качество чтений после чистки. Из рисунка видно, что исчезли чтения, у которых на концах были нуклеотиды плохого качества. Теперь нет позиции, в которой значение качества находится в красной области. Только одна позиция расположена в желтой. 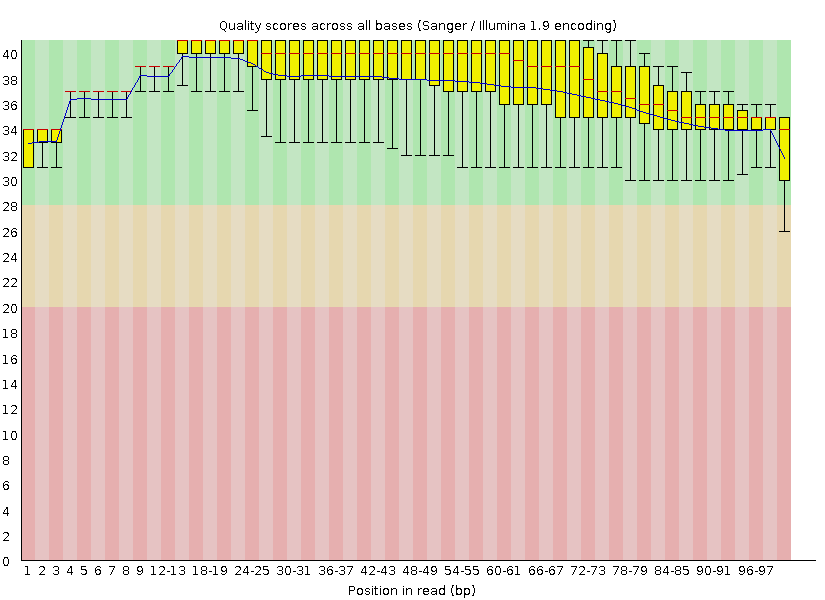
Также были удалены чтения длиной меньше 50 нуклеотидов (рис.5). 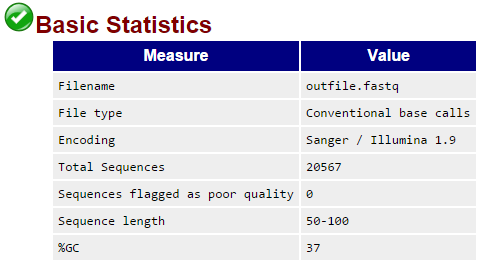
Часть II. Задание 3. Картирование чтений. Чтобы откартировать чтения, использовалась программа BWA. Сначала требовалось проиндексировать референсную последовательность с помощью команды bwa index (команда со всеми параметрами представлена в табл.1). Затем было построено выравнивание прочтений и референса в формате .sam. Для этого был использован алгоритм mem (табл.1). Задание 4. Анализ выравнивания. Для анализа выравниваний использовался пакет samtools. Сначала выравнивание чтений с референсом было переведено в бинарный формат .bam при помощи команды view. Далее нужно было отсортировать выравнивание чтений с референсом (получившийся после картирования .bam файл) по координате в референсе начала чтения (команда samtools sort). При помощи команды samtools index проиндексировала отсортированный .bam файл. Команда samtools idxstats позволяет узнать, сколько чтений откартировалось на геном (рис.6). Команды со всеми параметрами представлены в табл.1. 
Информация о картировании выдается в таком формате: название последовательности, длина последовательности, число картировавшихся ридов, число некартировавшихся ридов. Как видно из рис.6, картировалось 20566 ридов (хотя всего после чистки осталось 20567), некартировавшихся чтений не было. Часть III. Задание 5. Поиск snp и инделей. Для начала с помощью команды samtools mpileup -uf был создан файл с полиморфизмами в формате .bcf. Далее был получен файл со списком отличий между референсом и чтениями в формате .vcf. Для этого была использована команда bcftools call -cv пакета bcftools. Команды с параметрами приведены в табл.1. Всего было найдено 235 полиморфизмов, из них 17 инделей и 218 замен. На рис.7 представлена информация о трех полиформизмах. 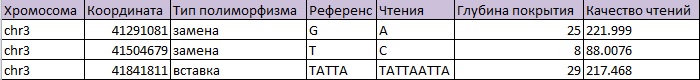
Задание 6. Аннотация snp. С помощью программы annovar нужно проаннотировать только полученные snp. Для этого нужно использовать базы данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar. Программа annovar установлена на kodomo: /nfs/srv/databases/annovar. Для работы с программой из .vcf файла необходимо было получить файл в формате avinput, с которым умеет работать эта программа. Файл был получен с помощью скрипта convert2annovar.pl (команда приведена в табл.1). Для аннотации файла с snp с помощью предложенных баз данных использовался скрипт annotate_variation.pl (команды для аннотирования по разным базам данных приведены в табл.1). При аннотировании с помощью базы RefGene было найдено 63 het и 155 hom замен. Из них 13 попали в экзоны, 4 - в UTR3 (3'-некодирующая область), 2 - в UTR5 (5'-некодирующая область), остальные (199) - в интроны. Замены попали в гены ULK4, GNL3, FNDC3B. Но экзоны были затронуты только у генов ULK4 и FNDC3B. Ген ULK4 кодирует серин-треониновую протеинкиназу ULK4, которая фосфорилирует белки. В гене обнаружено 10 замен, 8 из которых несинонимичны, т.е. произошла замена аминокислоты в белковой последовательности. Нуклеотидные и аминокислотные замены показаны на рис.7. Ген FNDC3B кодирует белок 3B, содержащий домен фибронектина III типа (может быть регулятором адипогенеза). В гене обнаружено 3 замены, 2 из которых являются синонимичными (замены аминокислоты не произошло, рис.7). 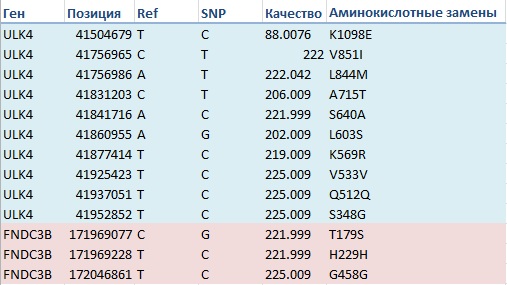
При аннотировании с помощью базы dbsnp были получены 2 файла: dbs.hg19_snp138_dropped и dbs.hg19_snp138_filtered. В первом файле представлены snp, имеющие snp. Из найденных snp rs имеют 179 штук. Во втором файле - snp, не имеющие rs (39 штук). Частота snp представлена в файле 1000g.hg19_ALL.sites.2014_10_dropped, полученом при аннотировании с помощью базы 1000 genomes. Как видно из данных частота полиморфизмов имеет довольно большой разброс: наименьшая частота полиморфизма 0.000599042, наибольшая - 0.996805. Клиническая аннотация snp приведена в файле gwas, полученном при аннотировании с помощью базы Gwas. Описывается 4 замены. Замена в гене ULK4 связана с кровяным давлением. Полиморфизм в гене GNL3 влияет на уровень гормона адипонектина, который участвует в регуляции уровня глюкозы и расщепления жирных кислот. Обе замены в гене FNDC3B влияют на рост. Файл с результатами аннотирования. Таблица 1. Команды, использованные в работе
|