Учебный сайт Полины Байкузиной | |||
| Главная | Семестры | О себе | Ссылки |
EMBOSSУпр.1. Требуется собрать несколько файлов в формате fasta в единый файл. 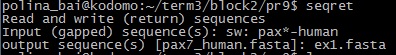
Результат: ex1.fasta Упр.2. Нужно разделить один файл в формате fasta с несколькими последовательностями на отдельные fasta файлы. Результат: 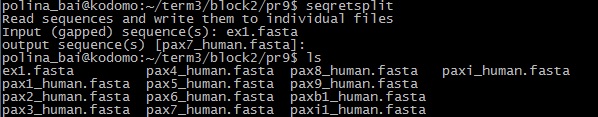
Упр.4. Транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода. Входной файл: rna.fasta 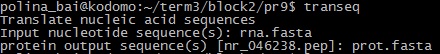
Результат: prot.fasta Упр.5. Транслировать данную нуклеотидную последовательность в шести рамках. Входной файл: rna.fasta 
Результат: protein.fasta Упр.6. Перевести выравнивание из fasta формата в формат .msf. Входной файл: align_09.fasta 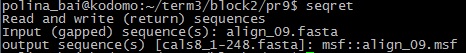
Результат: align_09.msf Упр.8. Перевести аннотации особенностей в записи формата .gb в табличный формат .gff. Входной файл:sequence.gb 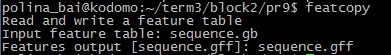
Результат: sequence.gff Упр.11. Найти частоты кодонов в данных кодирующих последовательностях. 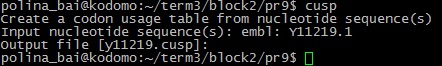
Результат: y11219.cusp Упр.12. Найти частоты динуклеотидов в данной нуклеотидной последовательности и сравнить их с ожидаемыми. 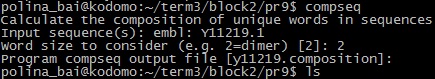
Результат: y11219.composition Сравннение аннотаций генов белков в хромосоме археи Desulfurococcus kamchatkensis с трансляциями длинных открытых рамок считывания. Для работы я выбрала архею Desulfurococcus kamchatkensis, с которой я работала в 1 семестре. 
Ее геном представлен одной кольцевой молекулой ДНК (NC_011766.1). Длина 1365223 п.н. Аннотация хромосомы. Нуклеотидная последовательность приведена в формате fasta. Задание 1. Получение списка трансляций открытых рамок с помощью команды getorf пакета EMBOSS. Требовалось выполнить команду со следующими опциями:
Команда: getorf -table 11 -minsize 180 -circular -find 0 1221.fasta out.fasta. Результат: out.fasta. Далее нужно было получить список координат и ориентаций найденных открытых рамок с помощью infoseq. Необходимые параметры:
Команда: infoseq -only -name -description -sprotein1 -length out.fasta > out.txt. Результат работы приведен в формате Excel. Задание 2.Список аннотированных генов белков. Аминокислотные последовательности всех белков представлены в файле NC_011766.faa. Список аннотированных белков был получен из файла NC_011766.ptt. Далее нужно было преобразовать таблицу в формат Excel (необходимые поля: locus_tag, from, to, ori, length, PID, product). Результат: таблица. Задание 3.Сравнение таблиц Excel. Далее нужно было сравнить две полученные таблицы. Для этого была сделана сводная таблица. Таблицы получились разными. Открытых рамок было найдено 5566, а аннотированных белков 1471, т.к. не каждая открытая рамка что-нибудь кодирует. На рис.2a, 2b, 3a, 3b показаны случаи, когда открытая рамка сдвинута на 3 нуклеотида, которые составляют один кодон. Из рис.2a и 2b видно, что последовательность открытой рамки заканчивается раньше, чем послеловательность гена. Возможно, программа getorf не учитывает стоп-кодоны в конце открытой рамки при указании координат рамки в геноме. На обратной цепи ситуация аналогичная (рис.3a и 3b). 



На рис.4а и 4b показаны примеры различий в длине аннотированного гена и открытой рамки: рамка длиннее гена. У бактерий помимо стандартного ATG есть и другие старт-кодоны: GTG, CTG, TTG, ATT. Из-за того, что открытая рамка начинается с нестандартного старт-кодона, открытые рамки оказались длиннее, чем гены. 

Не для всех аннотированных белков были получены открытые рамки считывания при помощи getorf, т.к. была установлена минимальная длина открытой рамки 180 п.н.(60 а.о.), а белки могут иметь длину около 30 а.о. (рис.5а и 5b). 

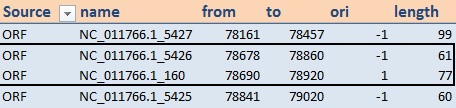
На рис.6а и 6b приведены примеры антипараллельных открытых рамок. На рис.6а представлен случай, когда на открытой рамке обратной цепи есть ген. Другая цепь не содержит ген. На рис.6b представлены антипараллельные открытые рамки, которые ничего не кодируют. 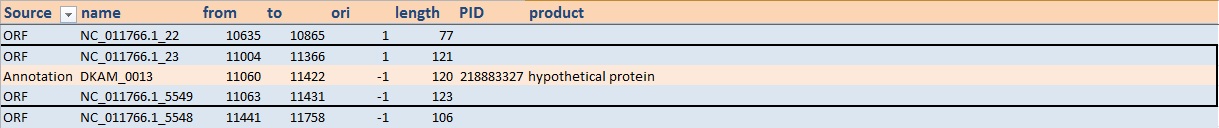
|