Учебный сайт Полины Байкузиной | |||
| Главная | Семестры | О себе | Ссылки |
Поиск сигналовЗадание 1. В данном задании требовалось сравнить состав систем рестрикции-модификации, закодированных в двух штаммах одного вида. 1. Сначала нужно было найти избегаемые сайты рестрикции в геноме выданной бактерии Acidaminococcus intestini (CP003058.1). Файл sites.list содержит список всех известных сайтов систем Р-М типа II. С помощью веб-сервиса было посчитано ожидаемое количество и контраст всех сайтов из списка в геноме бактерии (использовался метод Карлина). Выходной файл с веб-сервиса приведен здесь. Чтобы найти избегаемые сайты рестрикции, за порог было взято значение 0.78 (чтобы отличие от 1 можно было считать значительным). Всего было найдено 2 сайта, для которых контраст меньше 0.78. Файл с отобранными избегаемыми сайтами. 2. Далее нужно было найти избегаемые сайты рестрикции в наборе контигов из метагенома кишечника человека. Были выполнены те же шаги, что и в предыдущем пункте. В итоге был получен выходной файл с веб-сервиса. Был найдено 2 избегаемых сайта, файл с отобранными избегаемыми сайтами. 3. В обеих бактериях обнаружилось две системы рестрикции-модификации. И там и там было найдено 2 избегаемых сайта рестрикции (CTAG и GATC). Из соответствующей записи базы данных Nucleotide было выяснено, что бактерия, чей геном был секвенирован (пункт 1), обитала в перианальном абсцессе, а бактерия из пункта 2 - в кишечнике человека. Различий в числе и качестве систем модификации-рестрикции в данном случае не было обнаружено. Задание 2. В данном задании требовалось найти последовательности Шайн-Дальгарно (сайты инициации трансляции у прокариот) в геноме археи Desulfurococcus kamchatkensis 1221n. По данным статей, эта последовательность в общем случае составляет 5-6 нуклеотидов и расположена приблизительно в позициях -10 - -5 относительно старта трансляции [1]. Литературных данных о SD в геноме моей археи не нашлось. Для создания позиционной матрицы весов (PWM) с помощью MEME было выбрано около 500 хороших генов (длина больше 300 п.н., не гипотетические, хорошо аннотированы). Для них были получены последовательности от -15 до -1 нуклеотида (от начала CDS = старта трансляции). Для поиска SD по всему геному я получила последовательности для всех CDS от -20 до -1 нуклеотида. Далее был произведен запуск MEME со следующими параметрами: длина мотива от 4 до 6 п.н., поиск только по данной цепи (поскольку даны кодирующие последовательности), в последовательности ожидается от 0 до 1 появления мотива. В начале был проведен поиск до нахождения 3 мотивов, чтобы сравнить e-value (рис.1). Как видно из рисунка, e-value первого найденного мотива намного меньше, чем e-value второго и третьего. Затем был произведен запуск MEME для нахождения одного мотива и получения PWM для мотива SD. Найденный мотив (E-value: 1.6e-034) представлен на рис.2. 
Также я провела поиск, ограничив длину мотива 6 нуклеотидами. В найденном мотиве также явно выделяется GGTG (рис.3), но E-value в данном случае равняется 2.5e-039, поэтому далее PWM была получена для этого мотива. 
Позиционная матрица весов: Motif 1 position-specific probability matrix -------------------------------------------------------------------------------- letter-probability matrix: alength= 4 w= 6 nsites= 183 E= 2.5e-039 0.000000 0.000000 1.000000 0.000000 0.000000 0.000000 1.000000 0.000000 0.000000 0.000000 0.000000 1.000000 0.000000 0.000000 1.000000 0.000000 0.344262 0.114754 0.229508 0.311475 0.295082 0.098361 0.256831 0.349727 Затем проводился поиск по регионам перед всеми генами (от -30 до 1 нуклеотида, считая от старта трансляции) с помощью ресурса FIMO - всего 1470 генов. Запуск проводился с разными параметрами: при значении e-value < 0.01 было найдено 1137 мотивов, при 0.1 - более 5000, среди которых многие находки не относятся к SD. Для этих находок было построено распределение по началу относительно старта трансляции (рис.4). 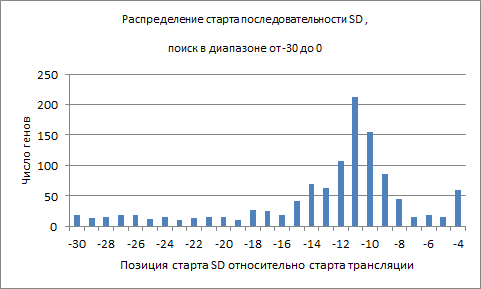
Как видно из гистограммы, находки в основном имеют начало в области от -10 до -12. На рис.5 представлен лого найденных сигналов. Процент генов, перед которыми найдена последовательность SD: 77%. Процент генов получился даже больше, чем бывает в реальности. Возможно, это связано с шириной участка, выбранного для поиска, Лого довольно хороший, поэтому есть вероятность, что результатам можно доверять. Задание 3. Нужно было определить сайты связывания данного транскрипционного фактора в данном участке хромосомы человека. Мне был выдан файл chipseq_chunk5.fastq с ридами Illumina. Сначала был сделан контроль качества прочтений с помощью программы FastQC (команда: fastqc chipseq_chunk5.fastq). На рис.6 представлены результаты работы программы FastQC, на котором показано качество определения нуклеотида в каждой позиции рида. 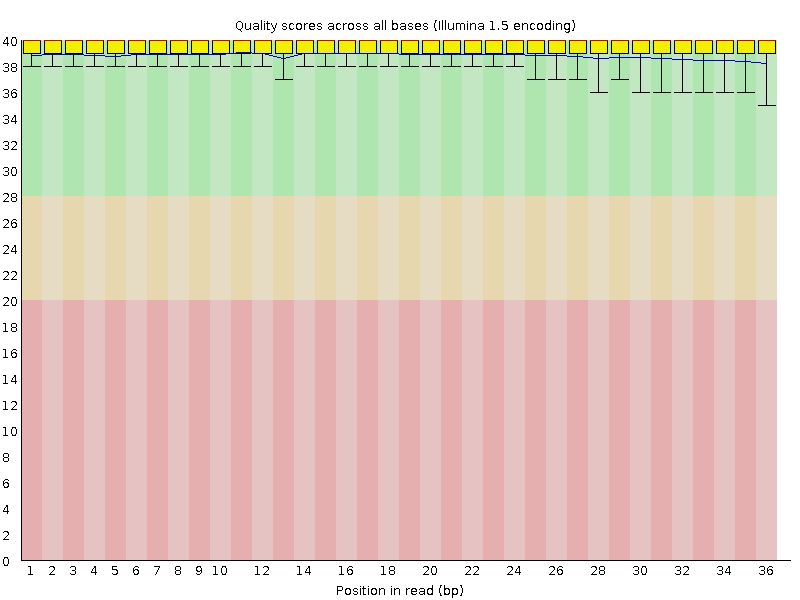
Поле графика разделено на 3 области (зеленый, желтый, красный), соответствующие качеству чтений (зеленый цвет - хорошее качество, красный - плохое). Как видно из рис.6 качество ридов очень хорошее (высокое значение качества). На рис.7 представлена основная информация о чтениях. Как видно из таблицы, количество чтений 6572. Длина чтений 36. 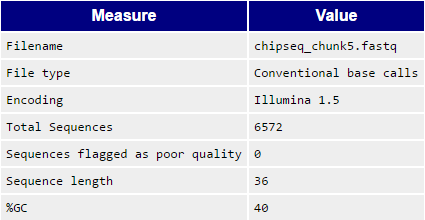
Так как качество ридов хорошее, отфильтровывать данные не пришлось. Далее с помощью программы BWA было выполнено картирование на геном человека hg19 (команда: bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk5.fastq > chipseq_chunk5.sam - построение выравнивания прочтений и референса в формате .sam; был использован проиндексированный файл). Для анализа выравниваний использовался пакет samtools:
Изначальное количество прочтений: 6572. Откартировалось на геном: 6572 . Для анализа были предложены прочтения с 12 хромосомы, так как больше всего прочтений откартировалось на нее (6185). Для поиска пиков использовалась программа MACS (команда: macs2 callpeak -t chipseq_chunk5.sorted.bam --nomodel -n my). В итоге было получено 3 файла: my_peaks.narrowPeak, my_peaks.xls, my_summits.bed. Все файлы содержат информацию о найденных пиках. Всего программой было найдено 9 пиков. Все они расположены в одном регионе 12 хромосомы - около 76 млн п.н. Ширина пиков варьируется от 213 до 497 нуклеотидов. Более подробная информация представлена в Табл.1. 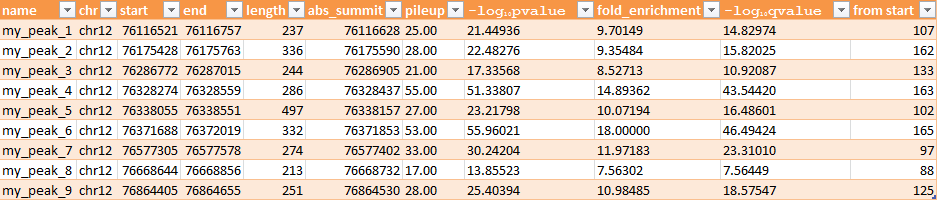
Информация была визуализирована с помощью браузера UCSC Genome Browser. Для этого в файл my_peaks.narrowPeak была дописана строчка: track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk X" browser position chr1:76116000-76865000. 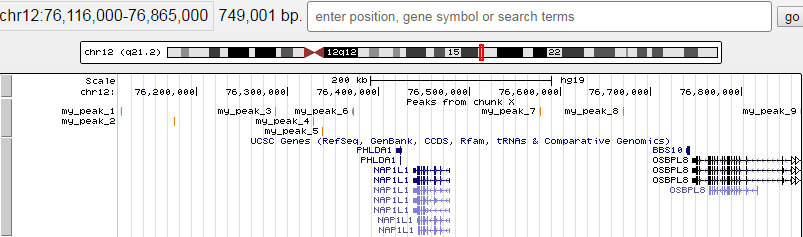
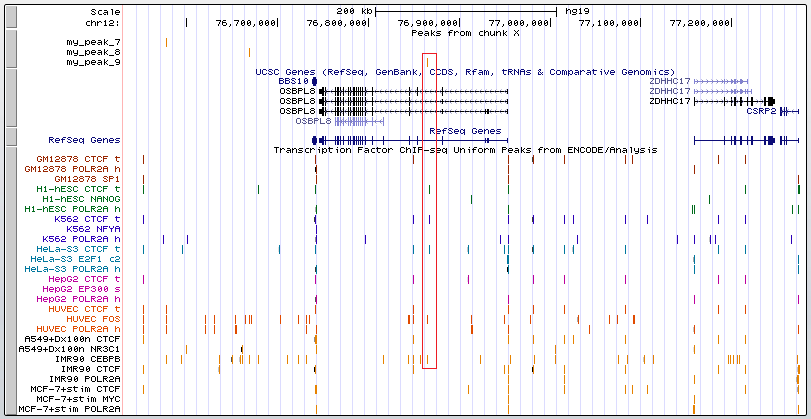
Расположение пиков: только один пик 9 попадает в область гена OSBPL8 (Oxysterol binding protein-like 8). Остальные пики с другими генами не перекрываются и расположены достаточно далеко от них. Ниже представлено более подробное описание пиков 2, 4, 9. 

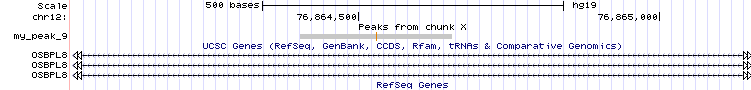
Для этих пиков я посмотрела их расположение относительно других факторов транскрипции из ранее проведенных ChIP-seq экспериментов. На рис.12-14 показаны в крупном масштабе пики 2, 4 и 9, соответственно, с хорошими значениями p-value и q-value, которые подтверждены результатами других экспериментов. 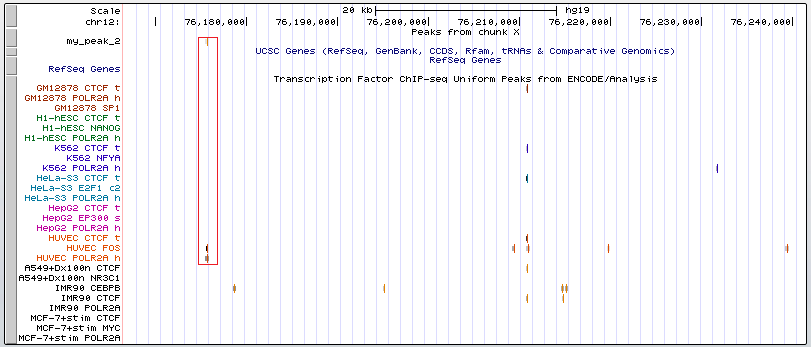
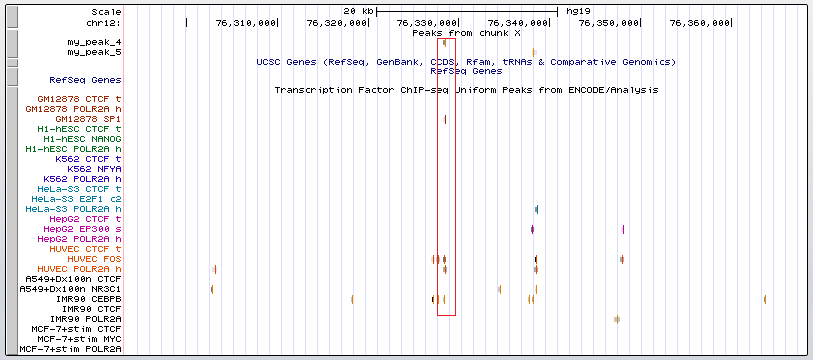
Задание 4. В данном задании требовалось проанализировать результаты ChIP-seq эксперимента по поиску ТАТА-box - сайтов связывания белка TBP. Это связывание необходимо и достаточно для инициации транскрипции у эукариот. ТАТА-бокс имеет консенсус T-A-T-A-A/T-A/G. В данном задании нужно было в геноме человека найти три гена, транскрипция которых инициируется с помощью TATA-бокс связывающего белка, и один - без сигнала TATA-бокса в промоторной области. Для анализа был выбран эксперимент на клеточной линии H1-hESC c помощью антител кролика. 
Результаты 1. Ген TCTA Название: Homo sapiens T-cell leukemia translocation altered gene Положение в геноме: chr3:49449639-49453909, + Координата старта транскрипции: 49449639 Длина гена: 4271 п.н. .png)
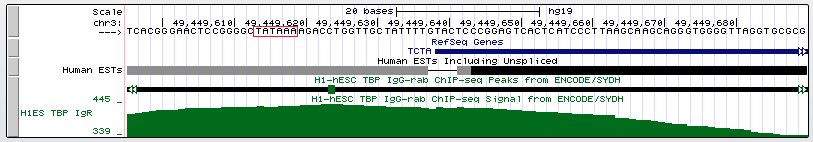
Найденный пик имеет довольно большую силу, что свидетельствует о его достоверности. Действительно, в промоторной области гена была найдена последовательность TATAAA примерно за 20 п.н. до старта транскрипции. 2. Ген C3orf14 Название: Homo sapiens chromosome 3 open reading frame 14, transcript variant 2 Положение в геноме: chr3:62304648-62321888, + Координата старта транскрипции: 62304648 Длина генома: 17241 п.н. .png)
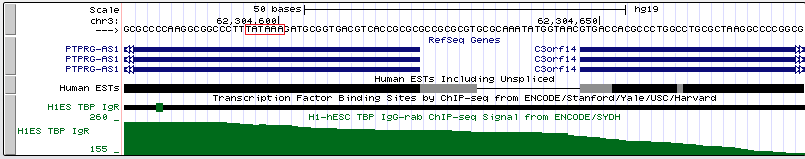
Был найден довольно хороший пик. Рядом была обнаружена последовательность TATAAA примерно за 30 п.н. до старта транскрипции. 3. Ген RPL24 Название: Homo sapiens ribosomal protein L24 Этот ген кодирует рибосомный белок, входящий в состав 60S субъединицы. Белок принадлежит к семейству L24E рибосомальных белков. Находится в цитоплазме. Положение в геноме: chr3:101399934-101405563, - Координата старта транскрипции: 101399934 Длина генома: 5630 п.н. .png)
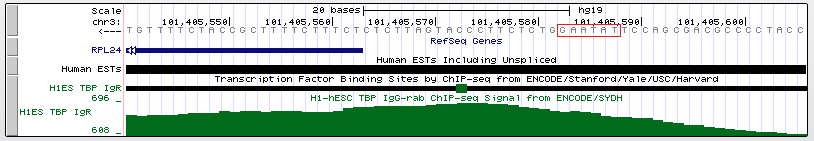
Пик связывания антител в промоторной области данного гена очень высокий (696, рис.20). Это свидетельствует о его высокой достоверности и, возможно, последовательности ТАТА-бокса, близкой к консенсусной. Действительно, примерно за 20 п.н. до старта транскрипции есть последовательность ТАТАAG. 4. Ген ALG1L2 Название: Homo sapiens ALG1, chitobiosyldiphosphodolichol beta-mannosyltransferase-like 2 Положение в геноме: chr3:129800674-129817233, + Координата старта транскрипции: 129800674 Длина генома: 16560 п.н. 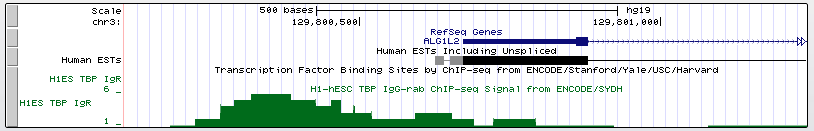
.png)
В промоторе этого гена нет ТАТА-бокса. Об этом свидетельствует отсутствие пиков в промоторной области и, собственно, отсутствие последовательности ТАТА-бокса в +/- 100 нуклеотидах от старта транскрипции. Использованные источники: [1] Wikipedia [2] Predicting Shine-Dalgarno sequence locations exposes genome annotation errors |