1. Построение дерева по нуклеотидным последовательностям
Для отобранных на предыдущем занятии бактерий требуется построить филогенетическое дерево, используя последовательности РНК малой субъединицы рибосомы (16S rRNA). Для этого сначала необходимо найти последовательности 16S рибосомальной РНК каждой из бактерий. Это можно сделать следующим способом. В записи EMBL, описывающей полный геном бактерии, найти соответствующее поле (FT с FTkey rRNA и упоминанием 16S rRNA в примечании). Полученные данные приведены в таблице 1:
| Название | Мнемоника | AC записи EMBL | Координаты РНК | Цепь |
| Bacillus subtilis | BACSU | AL009126 | 9810..11364 | + |
| Clostridium botulinum | CLOB1 | CP000726 | 9282..10783 | + |
| Clostridium tetani | CLOTE | AE015927 | 41801..43309 | - |
| Lactobacillus delbrueckii | LACDA | CR954253 | 45160..46720 | + |
| Listeria monocytogenes | LISMO | AL591981 | 99187..100732 | - |
| Staphylococcus epidermidis | STAES | AE015929 | 1722288..1723841 | - |
| Streptococcus aureus | STAA1 | AP009324 | 531922..533476 | + |
Нужные участки из записи EMBL были вырезаны командой seqret:
seqret embl:xxxxxxxx -sask
Последовательности были помещены в один fasta-файл rna.fasta. Названия последовательностей были отредактированы.
Для создания выравнивания отобранных белков сервере kodomo была запущена программа muscle с параметрами по умолчанию:
muscle -in rna.fasta -out alignment.fasta
Дерево, построенное по выравниванию 16s рРНК из заданных бактерий программой MEGA (Neigbour joining tree):
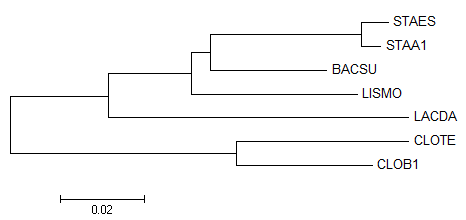 Рисунок 1. Neigbour joining tree
Рисунок 1. Neigbour joining tree
Как видно из сравнения с правильным деревом (рис 2.), полученное дерево имеет неправильную ветвь {STAES,STAA1,BACSU,LISMO} vs {LACDA,CLOTE,CLOB1}.
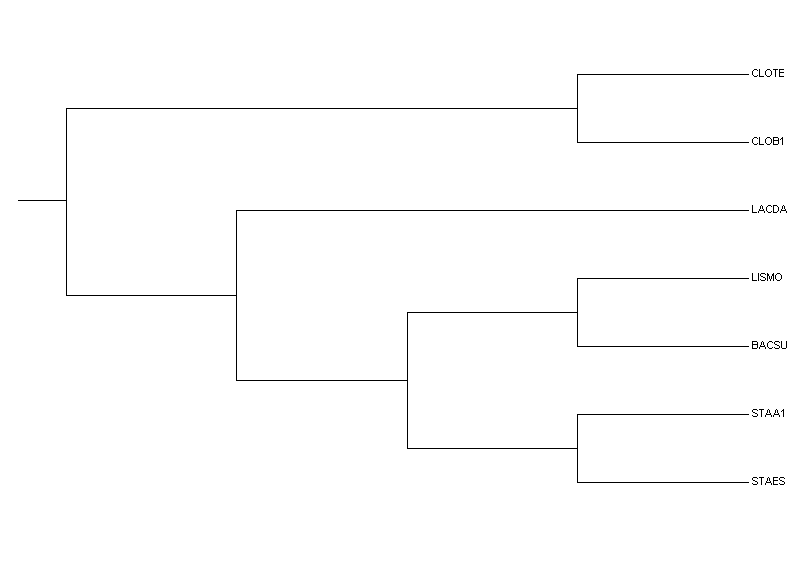 Рисунок 2. Правильное дерево
Рисунок 2. Правильное дерево
Для поиска гомологов предложен файл proteo.fasta, содержащий записи банка Uniprot, относящиеся к исходному списку бактерий. Поиск гомологов можно произвести с помощью программы blastp, а затем отобрать находки, относящиеся к отобранным бактериям. Последовательность команд для этого приведена ниже.
2. Построение и анализ дерева, содержащего паралоги
Для гомологов белка CLPX_BACSU в отобранных бактериях необходимо построить дерево.
Для поиска гомологов предложен файл proteo.fasta, содержащий записи банка Uniprot, относящиеся к исходному списку бактерий.
Поиск гомологов можно произвести с помощью программы blastp, а затем отобрать находки, относящиеся к отобранным бактериям.
Последовательность команд для этого приведена ниже:
seqret sw:clpx_bacsu
makeblastdb -in proteo.fasta -out proteo -dbtype prot
blastp -query clpx_bacsu.fasta -db proteo -evalue 0.001 -out clpx_blastp.out -outfmt 6
Нашлись такие гомологи из выбранных бактерий:
clpx_homologs_ids.txt
Были получены последовательности благодаря сервису retrieve на сайте UniProt.org
clpx_homologs.fasta.
На сервере последовательности kodomo были выровнены - файл clpx_homologs_a.fasta:
muscle -in clpx_homologs.fasta -out clpx_homologs_a.fasta
Дерево (рис. 3) построено, с помощью программу MEGA методом Neighbor-Joining.
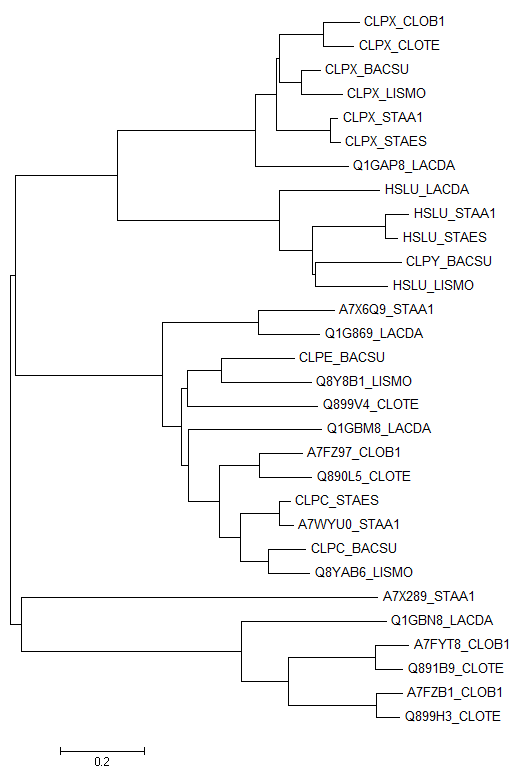 Рисунок 3. Дерево, содержащее паралоги
Рисунок 3. Дерево, содержащее паралоги
На построенном дереве ортологами являются, например, CLPX_CLOB1 и CLPX_CLOTE, HSLU_STAA1 и HSLU_STAES.
На основе реконструированного дерева можно сказать, что паралогами являются, например, Q899V4_CLOTE и CLPX_CLOTE, Q1GAP8_LACDA и HSLU_LACDA.