
We have to make a sequence alignment of 6 sequences of proteins from family HSP70. I used the program "seqret" (EMBOSS) to load these sequences of proteins in format FASTA. Then I merged into one file and made an alignment by Jalview.
Here You can see some information about proteins I used:
Table 1.
| Entry | Entry name | Protein name | Organism | Supergroup |
| Q8TQR2 | DNAK_METAC | Chaperone protein DnaK (HSP70) | Methanosarcina acetivorans (strain ATCC 35395 / DSM 2834 / JCM 12185 / C2A) | Archaea |
| Q18GZ4 | DNAK_HALWD | Chaperone protein DnaK (HSP70) | Haloquadratum walsbyi (strain DSM 16790 / HBSQ001) | Archaea |
| Q13E60 | DNAK_RHOPS | Chaperone protein DnaK (HSP70) | Rhodopseudomonas palustris (strain BisB5) | Bacteria |
| Q73GL7 | DNAK_WOLPM | Chaperone protein DnaK (HSP70) | Wolbachia pipientis wMel | Bacteria |
| Q74ZJ0 | HSP7F_ASHGO | Heat shock protein homolog SSE1 | Ashbya gossypii (strain ATCC 10895 / CBS 109.51 / FGSC 9923 / NRRL Y-1056) (Yeast) (Eremothecium gossypii) | Eukaryota |
| Q8SQR8 | HSP7F_ENCCU | Heat shock protein homolog SSE1 | Encephalitozoon cuniculi (strain GB-M1) (Microsporidian parasite) | Eukaryota |
The alignment was made by Jalview by program Tcoffee with Defaults, colored by ClustalX with Identity Threshold = 100%. There are labels (identity 80%, plurality 100%, gaps): C - positions which are conserved 80% or more, F - absolutely functionally conserved, G - positions with gaps. To see the whole alignment press here.
Picture 1. The part of alignment of 6 sequences of proteins from family HSP70 wis labels.
The datas about alignment was taken by program "infoalign" (EMBOSS).
Table 2. Data when the conserved 100%.
| Name | SeqLen | AlignLen | GapLen | % of GapLen | Ident | % of Ident | Similar | % of Similar |
| DNAK_METAC_1-617 | 617 | 847 | 230 | 27,15 | 31 | 3,66 | 0 | 0 |
| DNAK_HALWD_1-641 | 641 | 847 | 206 | 24,32 | 31 | 3,66 | 0 | 0 |
| DNAK_RHOPS_1-633 | 633 | 847 | 214 | 25,27 | 31 | 3,66 | 0 | 0 |
| DNAK_WOLPM_1-640 | 640 | 847 | 207 | 24,44 | 31 | 3,66 | 0 | 0 |
| HSP7F_ASHGO_1-697 | 697 | 847 | 150 | 17,71 | 31 | 3,66 | 0 | 0 |
| HSP7F_ENCCU_1-658 | 658 | 847 | 189 | 22,31 | 31 | 3,66 | 0 | 0 |
Table 3. Data when the conserved 70%.
| Name | SeqLen | AlignLen | GapLen | % of GapLen | Ident | % of Ident | Similar | % of Similar |
| DNAK_METAC_1-617 | 617 | 847 | 230 | 27,15 | 284 | 33,53 | 12 | 1,42 |
| DNAK_HALWD_1-641 | 641 | 847 | 206 | 24,32 | 282 | 33,29 | 9 | 1,06 |
| DNAK_RHOPS_1-633 | 633 | 847 | 214 | 25,27 | 294 | 34,71 | 4 | 0,47 |
| DNAK_WOLPM_1-640 | 640 | 847 | 207 | 24,44 | 296 | 34,49 | 4 | 0,47 |
| HSP7F_ASHGO_1-697 | 697 | 847 | 150 | 17,71 | 129 | 15,23 | 53 | 6,26 |
| HSP7F_ENCCU_1-658 | 658 | 847 | 189 | 22,31 | 91 | 10,74 | 57 | 6,73 |
Table 4. Data when the functionally conserved 100%. (calculate by python script with matrix BLOSUM62)
| Name | SeqLen | AlignLen | GapLen | % of GapLen | Ident | % of Ident | Similar | % of Similar |
| DNAK_METAC_1-617 | 617 | 847 | 230 | 27,15 | 31 | 3,66 | 110 | 12,00 |
| DNAK_HALWD_1-641 | 641 | 847 | 206 | 24,32 | 31 | 3,66 | 110 | 12,00 |
| DNAK_RHOPS_1-633 | 633 | 847 | 214 | 25,27 | 31 | 3,66 | 110 | 12,00 |
| DNAK_WOLPM_1-640 | 640 | 847 | 207 | 24,44 | 31 | 3,66 | 110 | 12,00 |
| HSP7F_ASHGO_1-697 | 697 | 847 | 150 | 17,71 | 31 | 3,66 | 110 | 12,00 |
| HSP7F_ENCCU_1-658 | 658 | 847 | 189 | 22,31 | 31 | 3,66 | 110 | 12,00 |
Table 5. Average values.
| AlignLen | GapLen | % | Identity | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| identity 100% | % | plurality 100% | % | identity 70% | % | |||||
| 847 | 199,33 | 23,53 | 31 | 3,66 | 246 | 29,04 | 31 | 3,66 | ||
I used a sequence of protein called Human Insulin. With help of "msbar" (EMBOSS) I have done 7 generations of mutant insulin (7 artificial point mutations in every generation), then I combined these mutant generations and the origin one in the file (see "Bash script").
Bash script (task 2)The alignment was made by Jalview by program Tcoffee with Defaults, colored by ClustalX with Identity Threshold = 100%. To see the whole alignment press on picture or here.
Picture 2. The alignment made by program.
Table 5. The list of artificial mutations in insulin.
| Position | Type of point mutation | Generation |
| 2 | insert of S | p3 -> p4 |
| 3 | deletion of A | p4 -> p5 |
| 9 | insert of N | p1 -> p2 |
| 10 | insert of L | p1 -> p2 |
| 11 | insert of L | p1 -> p2 |
| 12 | insert of L | p6 -> p7 |
| 21 | insert of N | origin -> p1 |
| 29 | deletion of A | p2 -> p3 |
| 29 | insert of A | p5 -> p6 |
| 30 | insert of Y | p2 -> p3 |
| 31 | insert of A | p2 -> p3 |
| 36 | insert of V | p5 -> p6 |
| 75 | replacement R to S | p3 -> p4 |
| 78 | replacement E to R | origin -> p1 |
Picture 3. The alignment made by program with some changes which I did.
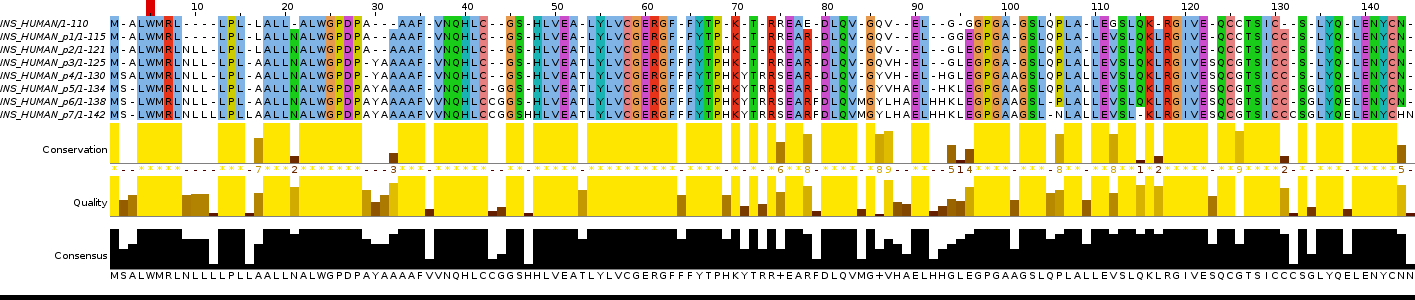
I had to change some items in the alignment (To see the whole alignment press here):
I used the nucleotide sequence of the gene of the protein called Human Insulin. With help of "msbar" (EMBOSS) I have done 7 generations of mutant insulin gene. Than with help "transeq" (EMBOSS) I made the transcriptions and constructed alignment of the respective mutant proteins (you can see the script). The alignment was made by Jalview by program Tcoffee with Defaults, colored by ClustalX with Identity Threshold = 60%.