В рабочую директорию /nfs/srv/databases/ngs/potapenko/pr12 были скопированы файлы из директории /P/y14/term3/block4/SNP/rnaseq_reads: файл chr22.1.fastq (одноконцевые чтения, полученные секвенированием РНК, первая реплика выбрана случайно) и файл gencode.v19.chr_patch_hapl_scaff.annotation.gtf (разметка человеческого генома по версии Gencode19 для сборки hg19), а также референсная последовательность 22ой хромосомы (использовалась в предыдущем практикуме).
Чтения были проанализированы программой fastqc, потом командой trimmomatic были удалены с концов буквы с качеством 19 (букв с качеством ниже не было, буквы с качеством 20 и более были оставлены). Были удалены риды длиной меньше 50. До чистки риды в файле имели длину от 25 до 51 нуклеотида, после чистки остались только риды длиной 50-51. Изначально в файле было 24294 ридов, после удаления коротких осталось 23459 (удалено 3.44 %).
fastqc chr22.1.fastq java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr22.1.fastq reads.fastq TRAILING:20 MINLEN:50 fastqc reads.fastq
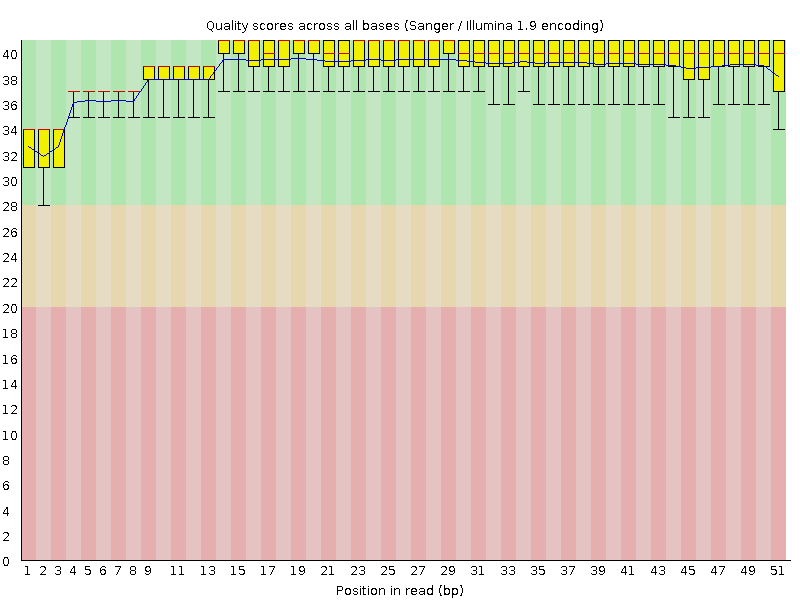
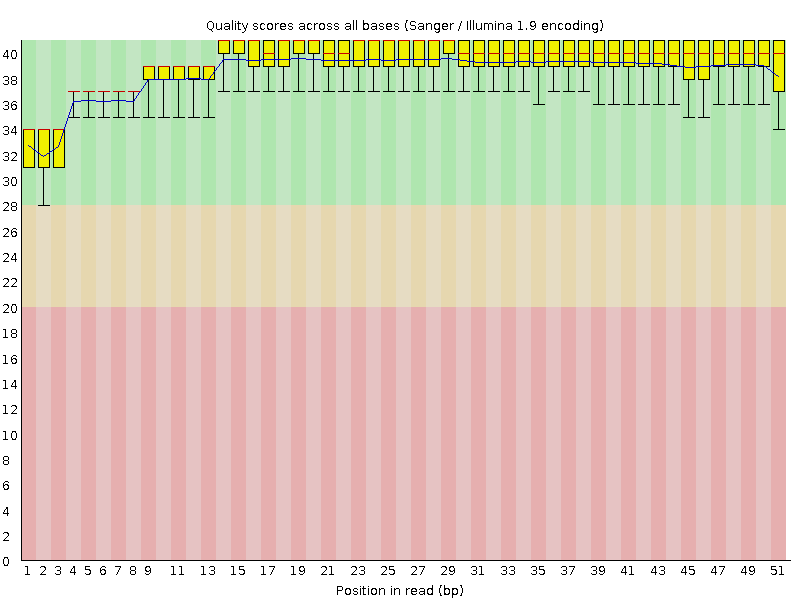
В выдаче trimmomatic график "FastQC "Per base quality"" практически не поменялся, т.к. букв с плохим качеством было очень мало. Видно, что качество ридов высокое, наиболее распространенное значение среднего качества букв в риде равно 39.
| | |
| Рисунок 1. Per base sequence quality до чистки. | Рисунок 2. Per base sequence quality после чистки. |
Для картирования ридов в рабочую директорию был скопирован файл chr22.fasta с референсной последовательностью 22ой хромосомы. Эта референсная последовательность была проиндексирована, в результате чего были созданы файлы chr22_indexed.1.ht2 - chr22_indexed.8.ht2:
hisat2-build chr22.fasta chr22_indexed
Можно было не индексировать референсную последовательность заново, а использовать ссылку на те же файлы, созданные в рабочей директории практикума 11. Далее риды были выровнены на референсную последовательность командой:
hisat2 --no-softclip -x chr22_indexed -U reads.fastq -S chr22.1_aligned.sam
Заметим, что программа hisat2 была запущена без параметра --no-spliced-alignment (в отличие от практикума 11), т. к. здесь нужно было выровнять риды РНК на ДНК, т. е. нужно допускать выравнивания с разрывами на интроны (которые есть в ДНК референса и которых может не быть в ридах РНК). В практикуме 11 выравнивались риды ДНК на референс ДНК, поэтому выравнивания с разрывом на интроны исключались. В результате этой команды было создан файл chr22.1_aligned.sam. На консоль была выведена следующая информация:
23459 reads; of these:
23459 (100.00%) were unpaired; of these:
366 (1.56%) aligned 0 times
23093 (98.44%) aligned exactly 1 time
0 (0.00%) aligned > 1 times
98.44% overall alignment rate
Видно, что есть невыровнявшиеся на референс риды (примерно полтора процента, это больше, чем для ридов, полученных секвенированием ДНК, в прошлом практикуме, но вряд ли их сравнивать), нет выровнявшихся больше одного раза.
Для дальнейшей обработке выравнивание на референс было переведено в бинарный формат:
samtools view -bS chr22.1_aligned.sam > chr22.1_aligned.bam
Риды в файле были упорядочены в соответствии с координатами участков референса, на которые они выровнялись:
samtools sort -T ./tmp/sorted.tmp -o chr22.1_sorted_alignment.bam -O bam chr22.1_aligned.bam
Файл с отсортированными ридами был проиндексирован:
samtools index chr22.1_sorted_alignment.bam
Чтобы разобраться с нужными опциями, help команды htseq-count был записан в отдельный файл:
htseq-count -h > htseq-count_help
Опция -f, она же --format, указывает на формат входного файла с выравниванием. Возможные значения - {sam, bam}, по умолчанию sam, в нашем случае .bam (т.к. проиндексированы файлы этого формата).
Опция -s, она же --stranded, указывает, на какую цепь референса выровнены риды Может принимать значения {yes,no,reverse}. Здесь yes означает прямую цепь, reverse - обратную цепь, no означает, что данные не специфичны к какой-то одной цепи. Нет основания предполагать, что риды РНК все относятся к какой-то одной цепи, поэтому значение 'no'.
Опция -i, или --idattr, указывает, какие строки GFF таблицы воспринимать как feature ID. По умолчанию gene_id, подходит под наш случай.
Опция -m, она же --mode, указывает, как программе обрабатывать риды, координаты выравниваний которых соответствуют более, чем одной feature. Множество возможных значений - {union,intersection-strict,intersection-nonempty}. Я выбрала union, чтобы узнать, если какой-то рид выровнялся на участки двух генов одновременно. При этом "теряется" информация о том, какие это два гена, но программа не так долго работает, если потребуется - можно запустить еще раз с теми же параметрами.
htseq-count -f bam -m union -s no -i gene_id chr22.1_sorted_alignment.bam gencode.v19.chr_patch_hapl_scaff.annotation.gtf > htseq-count_result
Результат работы программы записан в файл htseq-count_result. Этот файл содержит примерно столько же строк, сколько было в таблице .gtf, большая часть строк содержит название гена и цифру ноль, это значит, что ни один рид не выровнялся на этот ген.
__no_feature 1355 # 1355 ридов выровнялись на референс, но не попали в границы генов __ambiguous 0 # нет ридов, чье выравнивание пересекается более, чем с одним геном __too_low_aQual 0 # нет ридов низкого качества (т.к. опция -a не задана, берется ее значение по умолчанию (10), а все риды имеют качество выше) __not_aligned 366 # 366 ридов не выровнены на референс - это те 1.5%, которые не выровняла hisat2 __alignment_not_unique 0 # нет ридов, выровнявшихся на референс не однозначно
Чтобы увидеть те гены, на которые выровнялись какие-то риды, файл с результатами был обработан (здесь -v инвертирует работу grep, -w указывает искать только слова, т.е. подстроки, ограниченные пробельными символами или знаками препинания):
grep 0 htseq-count_result -v -w > short_result
В получившемся файле всего три строки:
ENSG00000185686.13 21738 __no_feature 1355 __not_aligned 366
Идентификатор ENSG00000185686.13 соответствует гену PRAME (preferentially expressed antigen in melanoma). Это ген белка-репрессора транскрипции, подавляющий сигнал при связывании ретиноевой кислоты тремя типами рецепторов ретиноевой кислоты. Предотвращает процессы, индуцируемые ретиноевой кислотой: дифференциацию, пролиферацию и апоптоз.
Вернуться на страницу семестра
© potapenko 2017-2018