Была создана рабочая директория, в нее был скопирован файл с картированными ридами, отсортированными соответственно их координате в геноме, в формате .bam:
cd /nfs/srv/databases/ngs/potapenko/ cp ./pr12/chr22.1_sorted_alignment.bam ./pr13
Далее файл с картированными отсортированными ридами был переведен в формат .bed (расшифровывается как Browser Extensible Data). По умолчанию команда выводит результат своей работы на консоль, этот результат работы был записан в файл:
bedtools bamtobed -i chr22.1_sorted_alignment.bam > chr22.1.bed
Формат .bam бинарный, формат .bed - текстовый. Полученный файл содержит 23094 строк, строки имеют вид:
chr22 22888095 22888146 D00795:16:C7BV0ACXX:5:2208:21047:101214 60 +
Т.е. в каждой строке сначала указана хромосома (везде chr22, т.к. изначально были взяты риды, полученные секвенированием РНК, транскрибированных с этой хромосомы), далее - координаты рида на хромосоме (помним, что все риды имеют дину 50 или 51) "+" означает положительную цепь (во строках стоит "+"), "60" в предпоследней колонке, видимо, score (везде стоит "60", в описании формата на сайте сказано, что score может иметь значение от 0 до 1000. Можно предположить, что score одинаковый во всех строках и значение такое небольшое, т.к. все риды имеют небольшую почти одинаковую длину (?))
Команда bedtools intersect была запущена с опцией -h, чтобы выбрать, как и с какими опциями ее запускать. Команда принимает на вход два файла, название одного записывается после опиции -а, название второго после -b, и ищет пересечения между двумя файлами с особенностями. По умолчанию файлы должны иметь формат .bed, но возможна и обработка файлов .bam (тогда нужна опция -abam). В нашем случае один файл - это картированные сортированные риды, второй - файл с разметкой генов по версии Gencode для генома человека сборки hg19. Опция -u позволяет не выводить информацию о записях разметки генома, никак не пересекающихся с ридами.
bedtools intersect -u -a /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed -b chr22.1.bed > chr22.1_intersect.bed
Создан файл chr22.1_intersect.bed, содержащий 197 строк вида:
chr22 22890123 22901768 protein_coding PRAME
Понятно, что первые три столбца - это номер хромосомы и координаты позиция в хромосоме, куда выравнялся рид. Последний столбец содержит название белка, закодированного в гене, с которым пересекается данный рид. Во всех строках, кроме двух последних, в этом столбце записано "PRAME" (это уже знакомый preferentially expressed antigen in melanoma). Значит, этот ген пересекается со 195 ридами. В двух последних строках в этом столбце записано "LL22NC03-63E9.3". Это неохарактеризованный локус, для которого известен один транскрипт (та некодирующая РНК, риды которой и были найдены при картировании). Вероятно, отсеквенировать хотели транскрипты PRAME, а LL22NC03-63E9.3 попали случайно, т.к. находятся в геноме рядом (см. рисунок 1).
Опция -с добавляет в последний столбец информацию, сколько для каждой фичи из первого файла (в данном случае - для каждого элемента разметки генома) с ней пересекается фич из второго файла (в данном случае - сколько ридов пересекается с данной разметкой генома). При этом нельзя применить одновременно опции -с и -u: -с считает, сколькими фичами из второго файла покрываются фичи из первого, -u позволяет выводить только те фичи из первого файла, которые покрыты какими-то фичами из второго. Поэтому опицю -u надо убрать и добавить grep, чтобы убрать строки с нулями:
bedtools intersect -c -a /P/y14/term3/block4/SNP/rnaseq_reads/gencode.genes.bed -b chr22.1.bed | grep 0 -v -w > chr22.1_c_grep.bed
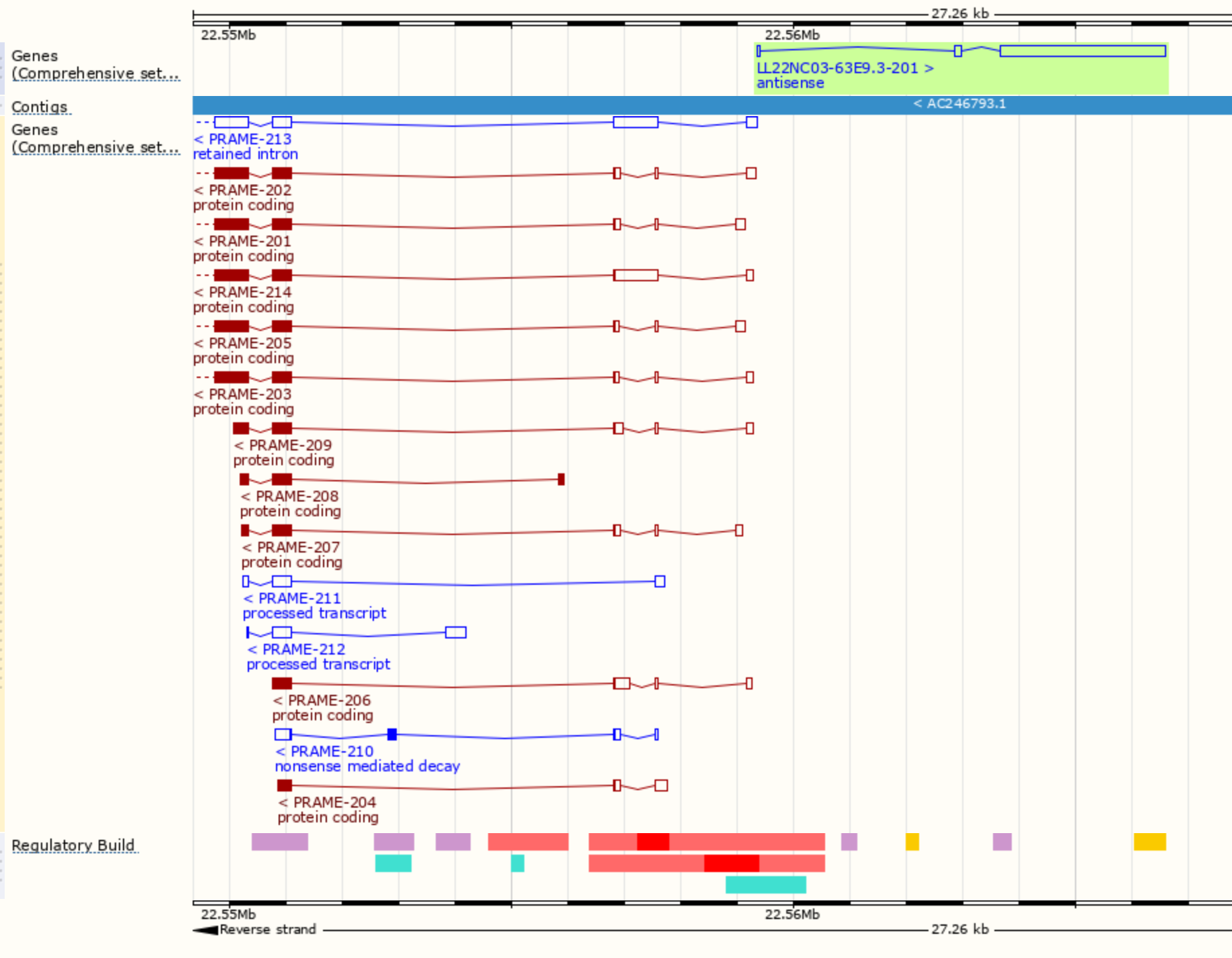
Рисунок 1. Белок-кодирующий ген PRAME и ген LL22NC03-63E9.3 лежат рядом на хромосоме, ген PRAME имеет 14 транскриптов (картинка с сайта ensemble).
Таблица 1. Характеристики генов, в границы которых попали риды.
| ген | PRAME | LL22NC03-63E9.3 |
| название | preferentially expressed antigen in melanoma | uncharacterized LOC648691 |
| размер гена | 11666bp | 7257bp (единственный транскрипт имеет длину 3108nt) |
| координаты | 22547696..22559361 | 22,559,343..22,566,599 |
| интронов | от 2 до 5 (у гена всего 14 транскриптов) | 2 |
| экзонов | всего 8, в разных транскриптах встречаются разные наборы | 3 |
| направление | reverse | forward |
Для поиска подходящих команд bedtools удобно использовать, например, "Summary of available tools" , где даны краткие описания команд. Чтобы узнать, какие опции нужны, использовался запуск команды с опцией -h.
1. Получите из файла c выравниванием файл с чтениями в формате fastq.
bedtools bamtofastq -i chr22.1_sorted_alignment.bam -fq chr22.1_task1.fastq
Замечу, что файл с ридами получен из бинарного файла с выравниванием (.bam), а не из файла в формате .bed, с которым работала команда bedtools intersect. Если нужно получить файл в формате .fastq из файла .bed, нужно сначала преобразовать его в .bam, а потом в .bed. При этом команда bedtools bedtobam будет использовать файл с разметкой генома.
2. Получите файл с нуклеотидной последовательностью (.fasta) для одного из покрытых Вашими чтениями генов.
bedtools getfasta -bed 1line.bed -fi ../pr11/chr22.fasta
На вход подаются два файла: fasta-файл с последовательностью, часть которой нужно вырезать, (этот файл использовался в практикуме 11, поэтому используется ссылка на другую директорию) и файл с координатами, на которые выровнялся один из ридов. Этот файл может быть в формате BED/GFF/VCF. Я вручную создавала файл 1line.bed, в который вырезала одну строку из файла chr22.1_intersect.bed. Важно следить, чтобы в файле с координатами колонки были разделены табуляцией (а не несколькими пробелами), иначе команда не работает. Записываемой в файл последовательности дается имя ">chr22:22899965-22900000", то есть имя состоит из номера хромосомы и координат по хромосоме.
Если в файле формата .bed будет несколько строк, то в fasta-файл запишутся нескольк последовательностей.
4. Объедините Ваши чтения в кластеры (используйте bed файл с выровненными чтениями из Обязательной части задания).
bedtools cluster -s -i chr22.1.bed > chr22.1_cluster_s.bed
На вход принимается файл с картированными ридами, отсортированными по координате. Если риды не отсортированы, команда выдает ошибку и указывает, например, что координаты в 4й строке меньше координат в 3ей. У команды есть две опции. Опция -s указввает, что нужно кластеризовать риды с учетом того, на какой цепи они находятся (по умолчанию направление ридов не учитывается). Опция -d указывает на максимальное расстояния между концами ридов, которые нужно лкастеризовать. По умолчанию стоит 0, т.е. кластеризуются риды, пересекающиеся или идущие стык в стык.
На выходе создается файл chr22.1_cluster.bed, каждая строка соответствует одному риду, в полсеней колонке указан номер кластера, к которому был отнесен данный рид. Всего кластеров 25 штук, из них первые 12 относятся к прямой цепи, остальные к обратной. Самый большой кластер - двенадцатый, к нему относится 22952 строк-ридов, координаты ридов в кластере лежат в пределах 22890117..22901634, т.е. в эти рамки попадают координаты ридов, откартировавшиеся на PRAME, так и откартировавшиеся на LL22NC03-63E9.3. Меня немного смущает, что при этом PRAME лежит на обратной цепи (?).
Много кластеров, содержащих совсем мало ридов. Даже есть кластеры, состоящие из одного рида (кластеры 21, 23, 24, 25). Это риды, которые лежат относительно далеко от других ридов. Если в команду добавить опцию '-d 10', кластеров будет всего 19, при этом явно какие-то кластеры в начале слились, поэтому самый большой кластер теперь имеет номер 8, а не 12. Отсюда понятно, что кластеры находятся недалеко друг от друга. Можно задать опцию '-d -10', т.е. разрешить кластеризовать кластеры, которые пересекаются не менее, чем по 10 нуклеотидам, тогда кластеров станет, наоборот, больше (логично).
Вернуться на страницу семестра
© potapenko 2017-2018