Код доступа к "моему" проекту по секвенированию - SRR4240360. На странице ebi.ac.uk/ena/data/view/SRR4240360 файл с чтениями был скачан в формате fastq по протоколу FTP (ссылка на скачивание во второй строке десятой колонки таблицы веб-страницы, под надписью 'FASTQ files (FTP)'.) Этот файл был перенесен в директорию /nfs/srv/databases/ngs/potapenko/pr14 и разархивирован командой:
gunzip --decompress SRR4240360.fastq.gz
При этом исходный файл был удалён и создан новый SRR4240360.fastq.
Для удаления адаптеров все адаптеры, лежащие в диреткории /P/y16/term3/block3/adapters, были перенесены в рабочую директорию (т.к. создавать файлы в той директории, где они лежат, у студентов нет доступа) и собраны в один файл. Команды:
cd /P/y16/term3/block3/adapters/ cp *.fa /nfs/srv/databases/ngs/potapenko/pr14 cd /nfs/srv/databases/ngs/potapenko/pr14 seqret '*.fa' adapters.fasta rm *.fa
Остатки адаптеров были удалены, качество ридов проанализировано до и псоле удаления концов:
fastqc SRR4240360.fastq java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 SRR4240360.fastq SRR4240360_trim_adapt.fastq ILLUMINACLIP:adapters.fasta:2:7:7 fastqc SRR4240360_trim_adapt.fastq
Были удалены буквы с концов. Удалялись буквы, качество которых ниже 20 (как в практикуме 11), и удалялись риды, которые после такого удаления букв стали короче 30 символов (в практикуме 11 этот параметр был 50, а не 30, но там и исходные риды были длиннее). Качество оставшихся ридов снова проанализировано. Выполненные команды:
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 SRR4240360_trim_adapt.fastq reads.fastq TRAILING:20 MINLEN:30
fastqc reads.fastq
Таблица 1. Изменения в файле с ридами при их подготовке. Видно, что в итоге ридов стало примерно на 10% меньше, качество немного восзросло после удаления плохих букв (их было двольно много на позициях с 30 и дальше. Видно, что было довольно много ридов, которые содержали длинные концы адаптеров и после обрезания адаптеров были удалены как слишком короткие. В исходном файле все риды имели длину 36, хотя в задании было сказано, что должно быть 39.
| - | всего ридов | длина ридов (интервал) | размер файла .fasrq |
| исходные риды | 8254632 | 36 | 832 М |
| после удаления адаптеров | 8212773 | 1-36 | 828 М |
| после удаления "плохих" концов и удаления коротких ридов | 7935084 | 30-36 | 798 М |
Применять для этого анализа программу fastq было излишне, вполне можно было посчитать число ридов grep-ом и каким-нибудь подобным способом посмотреть их длину.
velveth SRR4240360_kmers 29 -short -fastq reads.fastq
Длина к-меров была взята за 29 (так было указано в задании). Руководство к программе velveth (h на конце от слова hash) сообщает, что длина к-меров обязательносдолжна быть нечётной, иметь длину не более 31 основания (т.к. на хранение к-мера по умолчанию отводится 1 байт = 64 бит, а каждое основание - выбор из 4х возможных оснований, то есть два бита). Также длина к-меров должна быть меньше длины ридов, т.к. они должны быть подстроками ридов.
Создана папка SRR4240360_kmers, в ней три файла: файл Log (информация: когда программа была запущена, какой командой, версия программы...). Файл Sequences содержит последовательности длины 29 в формате fastq - все подпоследовательности ридов. Т.к. риды имеют длины 30, а подпоследовательности берутся длины 29, из каждого получается 2 подполседовательности. Но т.к. многие подпоследовательности встречаются во многих ридах, подпоследовательностей в файле Sequences намного меньше, чем число ридов * 2: действительно, файл с ридами весит 798 М, а файл Sequences - 579 М. Кажется, именно в этом заключается преимущество в объеме используемой памяти при работе с графами де Брайна.
Файл Roadmaps содержит строки вида 'ROADMAP n', где n - какое-то число, означающее, видимо, ссылку на к-мер (?).
velvetg SRR4240360_kmers/
velvetg была запущена со стандартными параметрами. Были созданы файлы: Graph, LastGraph, PreGraph, contigs.fa, stats.txt. Файл contigs.fa соерижит, как можно догадаться, контиги, подписанные так: '>NODE_454_length_66_cov_2.409091', то есть для каждого контига его название включает его длину и среднее покрытие ридами. Можно заметить, что номер (454) - это не есть номер контига в списке контигов, т.к. максимальный такой номер равен 1135, а контигов всего 505. Контиги упорядочены не по длине, но наиболее длинные находятся в начале списка.
Также файл Log был дописан (раньше там была информация о запуске velveth, теперь в конец добавилась информация о запуске velvetg ). Из него можно узнать, что N50 = 67095 (то есть не менее, чем половина генома покрыта контигами длины не меньше, чем это число), самый длинный контиг имеет длину 94956, суммарная длина контигов 707414.
Из файла stats.txt (содержащего, логично, статистику о контигах) можно узнать, что самый длинный контиг (94956) имеет среднее покрытие 43.7, а следующие по длине контиги имеют длины 70305 (покрытие 49.4) и 70300 (покрытие 42.0). Здесь и далее данные файла stats.txt обрабаотывались с помощью LibreOffice Calc (аналог Excel).
Медиана среднего покрытия для контигов - 4.8, среднее значение среднего покрытия - 12.5. Такие невысокие значения получаются из-за довольно большого числа контигов небольшой длины (в т.ч. длины 29). Понятно, что концы каждого контига имеют низкое покрытие позиций: если бы покрытие крайних позиция было бы высокое, эти концы содержались бы в еще каких-то ридах и это был бы не окнец контига, т.к. его можно было бы "продолжить". Поэтому именно короткие контиги "занижают" среднее покрытие. Так, 100 наиболее длинных контигов имеют среднее значение среднего покрытия 20.
Максимальное среднее покрытие контига - 116, но этот контиг имеет длину всего 37bp. Ничего с помощью алгоритма megablast, ограниченного таксоном Buchnera aphidicola, найдено не было. Зато контиг длиной больше 2000 bp и покрытием 97 (ID = 22) дает много находок (поиск аналогичный). В том числе нашлась плазмида, содержащая гены большой и малой субъединиц антранилат синтазы, а также эти же гены в составе хромосомы. Возможно, покрытие большое из-за того, что у этого биообразца Buchnera aphidicola эта последовательность есть и на плазмиде, и в основном геноме, может, даже есть несколько копий этой плазмиды.
Другой контиг с аномально высоким покрытием (100) имеет длину 502. Он выравнивается (100% Ident) на гены тех же двух субъединиц на хромосоме, чуть хуже (Ident 98.5%) на них же на плазмиде.
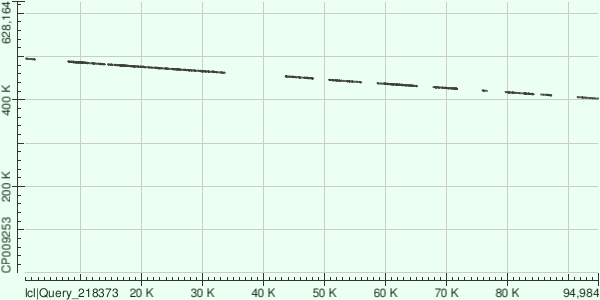
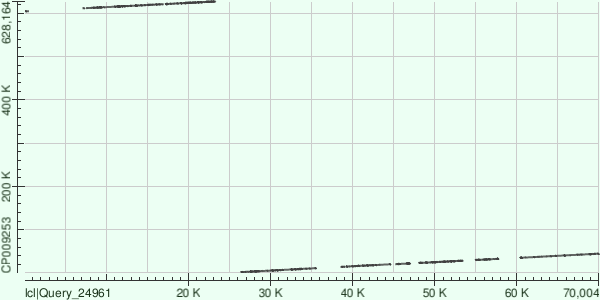
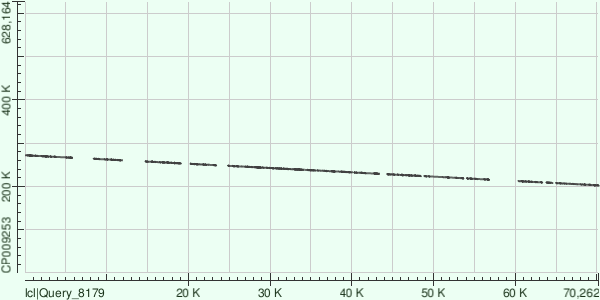
Таблица 2. Последовательности трех самых длинных контигов были скопированы в отдельные файлы, потом каждая из них была выровнена с хромосомой Buchnera aphidicola (AC — CP009253) алгоритмом megablast. Анализировались карты локального сходства и таблицы с характеристиками выравниваний, скачанные в формате .csv. На картах локального сходства по оси абсцисс отложен контиг, по оси ординат - референсная хромомсома.
| |
Первый по длине контиг (94956bp) имеет покрытие 43.7. Для этого контига было построено 20 выравниваний с разными участками генома. Т.е 20 кусочков этого контига выровнялись на 20 кусочков референса, а между этими кусочками в контиге лежат последовательности, не выровнявшиеся ни на какой участок референса. Видно, например, что среди этих 20 нет выравнивания начального участка контига (до 865 нуклеотида включительно). По Query cover = 60 % (цифра взята из таблицы, которую выдает BLAST как описание всех найденных выравниваний) можно судить, что эти невыровненные участки имеют довольно значительную длину. Видно, что цепи контига и референса противоположно напралены (иначе говоря, контиг лёг на обратную цепь референса). Контиг соответствует небольшому участку референса с координатами с 495148 по 402668. Средний % identity на этих выравниваниях - 77.3 % (посчитано как (сумма (identity * длину выравнивания)) / сумму длин выравниваний.) Такая доля контига, участвующая в выравнивании, и такой процент identity кажутся мне невысокими, т.к. предполагается, что это выравнивание последовательностей бактерий одного вида, а контиг при секвенировании имел высокое среднее покрытие. Можно заметить, что выравнивание контига с аномально высоким покрытием, построенное выше, имело 100% identity. |
| |
Второй по длине контиг имеет длину 70305 и среднее покрытие примерно 49.4 (его номер в списке контигов - 5). Для него построилось 13 выравниваний. Как видно по карте локального сходства и по координатам, цепи референсной хромосомы и контига сонаправленыю Координаты в геноме - с 2004 по 44693 и с 604302 по 627104. Это означает, что контиг ложится на то место в геноме, где координаты "закольцовываются" (геном кольцевой, но нужно какая-то позиция, с которой начинать нумерацию). Эти координаты, как и координаты для первого контига, не учитывают невыравненных участков между этими 13ю выравниваниями. Средний процент identity здесь 79.1 % (выше, но незначительно), доля контига, участвующая в выравнивании, тоже выше: 71%. |
| |
Для третьего по длине контига (длина 70300 bp, покрытие примерно 42.0, ID в списке контигов 9) построено 14 выравниваний. Ложится на обратную цепь референсной хромосомы. Самое длинное выравнивание имеет длину почти 11 тысяч bp, это больше 15 % длины контига. Это больше, чем у первого контига и по процентному, и по абсолютному значению (у первого самое длинное выравниваение - 7389 bp), у второго контига все выравнивания были еще короче. Все выравнивания соответствуют координатам хромосомы с 202390 до 271926 (как и раньше, не учитывая разрывы). Средняя identity для этих покрытий 76.9%, доля контига, участвующая в выравнивании - 79% (выше, чем у первых двух контигов). |
Вернуться на страницу семестра
© potapenko 2017-2018