В этом задании предлагается сравнить выдачи двух программ, предсказывающих вторичную структуры нуклеиновых кислот, между собой и с работой find_pair.
Программа einverted (пакет emboss) при стандартных параметрах не находит инвертированных повторов в последовательности тРНК (глутаминовая тРНК из практикума 2, pdb_id = '1gts'). Если снизить параметр minimum score threshold, не меняя при этом других параметров, программа находит акцепторный стебель (score = 18) (см. таблицу 1). Если, кроме того, снизить штрафы за гэпы, программа находит, кроме акцепторного, антикодоновый стебель. Довольно логично, что для этого нужно снизить штраф за разрыв, ведь расстояние между этими двумя плечами разное по двум цепям. У меня не получилось настроить параметры программы так, чтобы она находила какой-то другой стебель. По крайней мере, если уменьшать штрафы за гэпы и за неспаренные основания, программа выдает связи там, где в JMol основания совсем далеко друг от друга, но не находит другие стебли. Видимо, это связано с алгоритмом программы.
Можно предположить, что программа находит не все стебли потому, что она ищет только канонические пары, а во вторичной структуре тРНК много неканонических, как было выяснено в предыдущем практикуме.
Программа RNAfold также может быть использована для предсказания вторичной структуры РНК. Она выдает вторичную структуру в форме правильной (?) скобочной последовательности, а также в виде карты контактов нуклеотидов и в виде плоского рисунка предсказанной структуры (для преобразования фалов .ps использовалась ps2pdf). Программа сразу нашла все четыре стебля (при стандартных настройках).
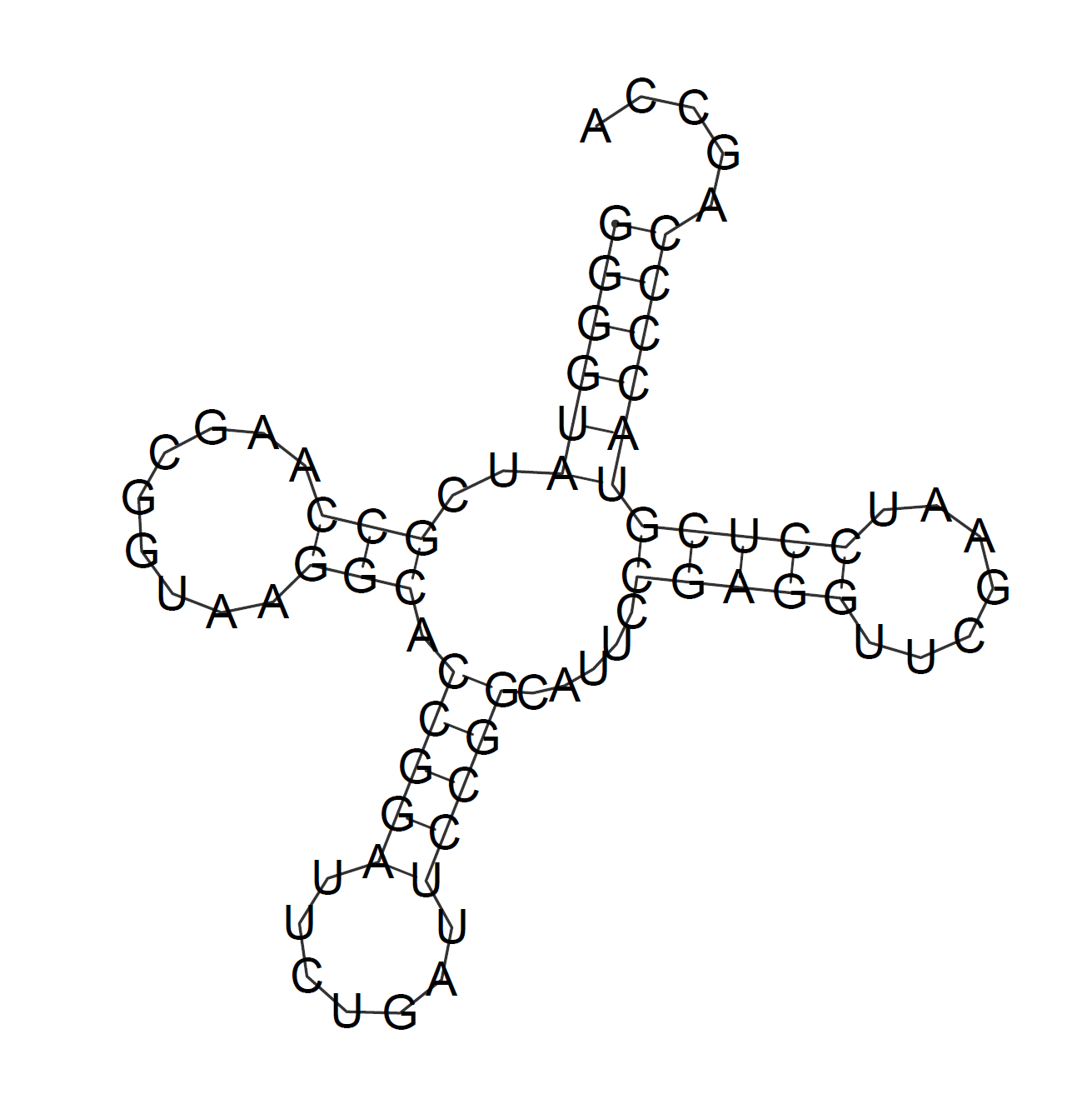
Рисунок 1. Вторичная структура тРНК, предсказанная RNAfold.
В этой таблице представлено сравнение работы программ, предсказывающих вторичнцю структуру РНК. Подсчитаны только канонические пары оснований, образующие стебли. Программы einverted и RNAfold не могут найти неканонические пары, спаренные основания не в составе стебля также не были найдены. Программа find_pair может находить как неканонические пары, так и одиночные пары.
В файле pdb старого формата (и, соответственно, в выдаче find_pair тоже) основания нумеруются с 2 (у меня не получилось понять, почему так), в программах, работавших с последовательностью, нумерация с 1. Это учитывалось в сравнении результатов работы программ.
Таблица 1. Вторичная структура тРНК,предсказанная разными способами.
| участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5' - 2-7 - 3'; 5' - 66-71 - 3'; всего 6 канонических пар | найдены все 6 канонических пар | найдены все 6 канонических пар |
| D-стебель | 5' - 10-12 - 3'; 5' - 25-23 - 3'; всего 3 канонических пары | стебель не найден | найдены все 3 канонические пары |
| T-стебель | 5' - 49-54 - 3'; 5' - 65-61 - 3'; всего 5 канонических пар | стебель не найден | найдены все 5 канонических пар |
| Антикодоновый стебель | 5' - 37-44 - 3'; 5' - 33-26 - 3'; всего 8 пар, из них 5 канонических. | найдены все 5 канонических пар | найдены все 5 канонических пар |
| Общее число канонических пар нуклеотидов | всего 22 пары, из них 19 канонические | 11 из 19 канонических | найдены все канонические пары |
Выбранная структура имеет pdb_id = 1pp8; рассматриваются только цепи M, R, Y и ДНК.
Ссылка на скрипт для JMol. В начале скрипта опредлены с помощью define необходимые множества атомов, потом они поочередно выделяются в структуре.
Как определялись необходимые множества атомов, можно посмотреть здесь
Пример того, как я искала контакты, для случая полярных контактов между белком и остатками фосфорной кислоты в ДНК: $ select within(3.5, o_phosphate) and prot_polar 8 атомов выбрано $ color green; cpk 200 $ select within(3.5, prot_polar) and o_phosphate 7 атомов выбрано $ color violet; cpk 200
Здесь prot_polar - полярные атомы рассматриваемых цепей белка, o_phosphate - кислороды в остатках фосфорной кислоты в ДНК. После того, как атомы, предположительно участвующие в контакте, выделены, можно уточнить их число. Например, здесь со стороны белка в контактах участвуют 8 атомов, а со стороны ДНК - 7 атомов. В JMol видно, что это из-за того, что один из кислородов со стороны ДНК контактирует одновременно с двумя атомами со стороны белка. JMol может посчитать число атомов, находящихся на заданном расстоянии от другой группы атомов. При этом один атом из группы может контактировать с несколькими атомами из другой группы, это приходится смотреть вручную. Результаты приведены в таблице 2.
Таблица 2. Контакты разного типа в комплексе XXXX.pdb
| Контакты атомов белка с: | Полярные | Неполярные | Всего контактов |
| остатками 2'-дезоксирибозы | 1 | 24 контакта (13+13 атомов) | 25 |
| остатками фосфорной кислоты | 8 | 13 контактов (13+5 атомов) | 21 |
| остатками азотистых оснований со стороны большой бороздки | 0 | 7 контактов (5+4 атомов) | 7 |
| остатками азотистых оснований со стороны малой бороздки | 16 контактов (13+6 атомов) | 0 | 16 |
Видно, что неполярных контактов больше, чем полярных; в том числе много контактов за счет атомов углерода в дезоксирибозе. Хотя, возможно, это только за счёт того, что среди атомов углерода в дезоксирибозе 3 легко доступных для контакта с белком. Эти контакты неспецифичные к последовательности.
Получить схему контактов ДНК-белок для структуры с pdb_id = 1pp8 с помощью nucplot у меня не получилось (программа не создает файл со схемой контактов, хотя создает некоторые другие файлы). Поэтому я взяла другую структуру с pdb_id = 1mnm.
get-pdb 1mnm remediator --old "1mnm.pdb" > "1mnm_old.pdb" nucplot 1mnm_old.pdb ps2pdf nucplot.ps
Последняя команда использовалась для перевода файла .ps в .pdf. Сначала я попыталась использовать convert, но качество картинки было заметно хуже.
Pdf-файл с полученной схемой контактов.
Большинство представленных на схеме аминокислотных остатков имеют всего один или два контакта с ДНК (с одним атомом ДНК, когда на схеме связи указаны параллельными линиями, или с разными, когда линиии связей идут в разные стороны).. Многие остатки, образующие несколько контактов, указаны звездочкой, т.к. подписаны на схеме дважды. На схеме нет остатков, имеющих больше трех контактов с ДНК, но остатков, имеющих три контакта с ДНК несколько. Например, гистидин-134 в цепи D (см. рисунок 2). Также на схеме есть остаток фосфорной кислоты в составе ДНК, образующий связи с 4 аминокислотными остатками.
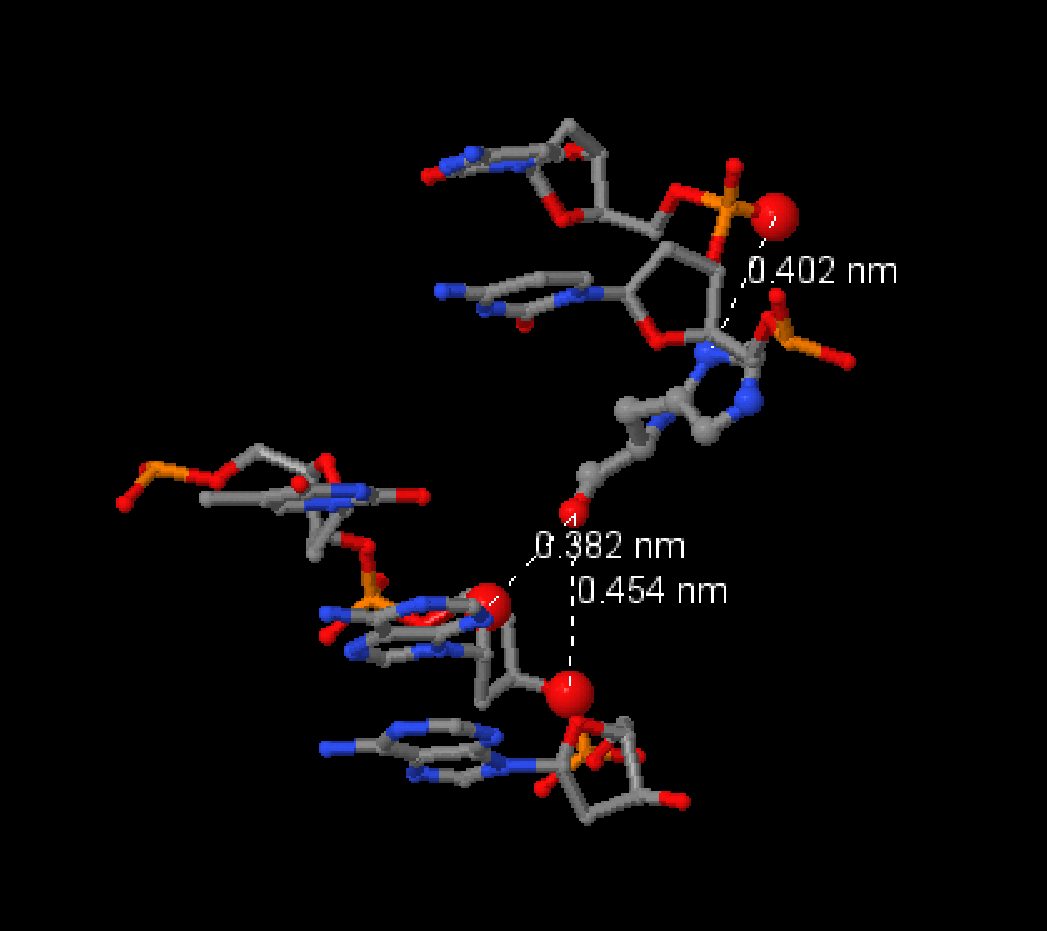
Рисунок 2. Аминокислотный остаток [hys]134:D образует 3 водородные связи: одну с остатком фосфорной кислоты нуклеотида [DC]7:E и две с остатком сахара нуклеотида [DA]50:F (т.е. связан с двумя цепями ДНК одновременно). Правда, расстояния между электроотрицательными атомами, образующими связи, больше принятых для водородной связи длины в 3.5А. Но связей несколько, поэтому суммарно взаимодействие не такое слабое.
Важными для узнавания последовательности ДНК белком являются аминокислотные остатки, образующие контакты с азотистыми основаниями. Здесь мне кажется наиболее важным остаток лизина-38. Он образует (по результатам nucplot) водородные связи с соседними азотистыми основаниями (см. рисунок 3).
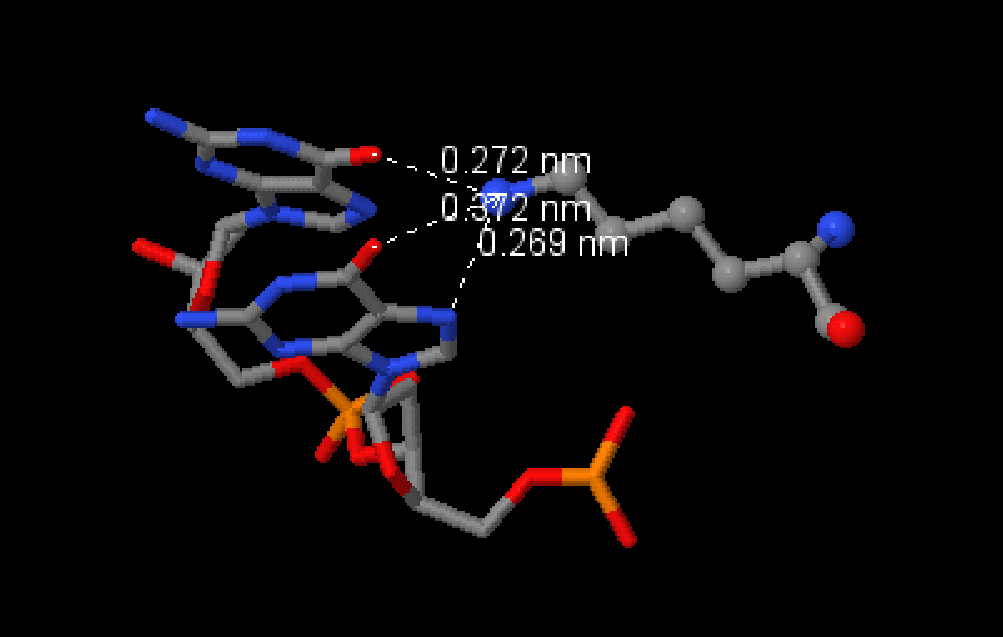
Рисунок 3. Аминокислотный остаток [lys]38:B образует водородные связи с основаниями [DG]14-15.
Вернуться на страницу семестра
© potapenko 2017-2018