Консенсусная последовательность , полученная по выравниванию из практикума 6 программой consambig пакета EMBOSS. Большими буквами программа выделила часть последовательности, которая подтверждена и прямой, и обратной цепью.
Для поиска похожих последовательностей использовался алгоритм BLASTN, т.к. не было предположения, что нашедшиеся последовательности будут очень похожи. Поиск производился по банку Nucleotide collection (nr). Вот лучшие находки:

Первая находка отличается от последовательности, по которой производился поиск, всего на 4 нуклеотида, если не считать того, что она не содержит начальный участок "моей" последовательности длиной 21 нуклеотид, причем одно из этих 4 отличий - это тот символ, который нельзя было однозначно распознать на хроматограмме и обозначила R (A или G). Следующие несколько находок отличаются в тех же позициях.
Все находки, кроме одной, относятся к cytochrome oxidase subunit I (CO1). Правда, в разных находках номер субъединицы обозначен либо римской, либо арабской цифрой, но это один и тот же ген. Большинство находок, в том числе вся верхняя половина списка, относится к митохондриальному геному вида Ophiopholis aculeata (это вид морской звезды, встречается в том числе в Белом море). В этом виде есть 4 подвида, но на страницах найденных последовательностей не указано, к какому подвиду они принадлежат. Шифр, идущий после слов isolate или voucher - это номер образца. Можно считать, что последовательность, по которой производился поиск, содержит (начиная со свеого 22го нуклеотида) ген первой субъединизы цитохром оксидазы и принадлежит тому же виду или очень близкому виду.
Цитохром оксидаза - это IV комплекс в дыхательной цепи переноса электронов. Она относится к суперсемейству гем-медных оксидоредуктаз, для которых характерна консервативность первой субъединицы ( Jiapeng Zhu, Kutti R. Vinothkumar & Judy Hirst. Structure of mammalian respiratory complex I, найдено через статью на elementy.ru) Эта консервативность и видна в поиске: первые 46 находок имеют E-value = 0. По консервативным последовательностям хорошо искать дальних гомологов, отличить по консервативной последовательности близкие виды одного рода не всегда возможно.
Так как последовательности оказались очень похожи, я решила повторить поиск по той же базе данных, но с алгоритмом megablast и ограничив поиск таксоном Ophiopholis. Лучшие находки оказались все те же, что и в первом поиске. Ой, это уже следующее задание.
В этом задании три разных варианта - это предложенные в условии megablast, blastn с параметрами по умолчанию, blastn с чувствительными параметрами. Как оказалось, последовательность с выданной мне хроматограммы выравнивается с Е = 0 на белки организма Ophiopholis aculeata. Так как между выровненными последовательностями различия очень небольшие, разницы между работами алгоритмов не видно совсем (проверено). Поэтому, чтобы поиск был сколько-нибудь осмысленным, я исключила из рассмотрения вид Ophiopholis aculeata, но ограничила родом Ophiopholis (чтобы не ждать долго и заодно узнать, кто еще есть в этом таксоне). Для этого при поиске я сначала ограничила род, потом добавила еще одно условие, ввела название вида и нажала "исключить". Вот что у меня получилось:
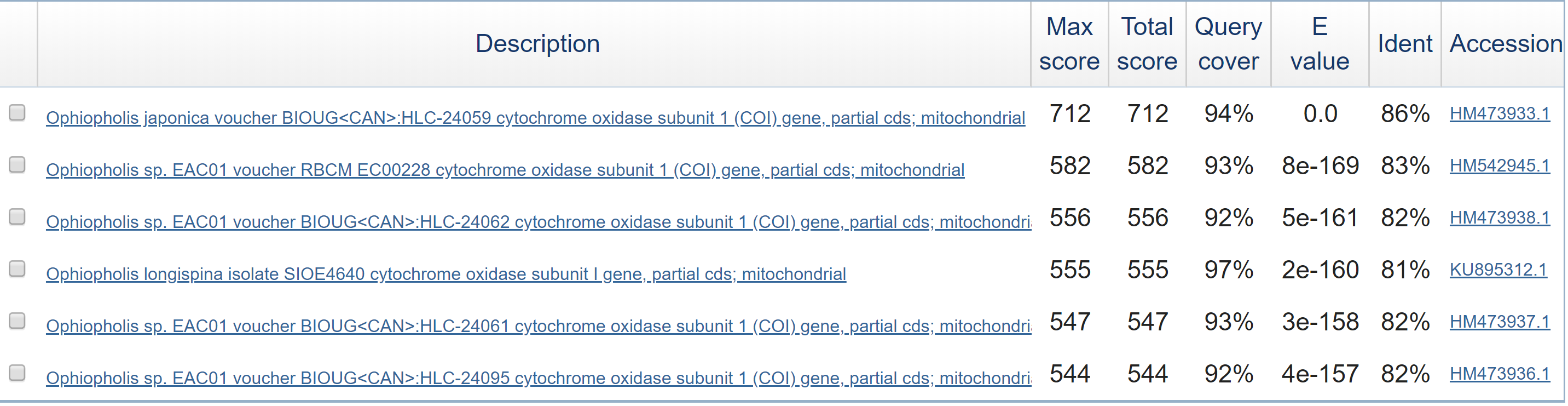
Рисунок 2. поиск megablast (последовательность с хроматограммы, исключен вид). Находок мало (6, все на картинке). Дело в том, что этот алгоритм приспособлен искать очень похожие последовательности. Зато узнали, у какого другого вида наиболее похожая цитохром оксидаза. Что интересно, у первой находки Е написано 0 (как я понимаю, выдается ноль, если меньше 1е-200). А ведь это всё-таки другой вид!
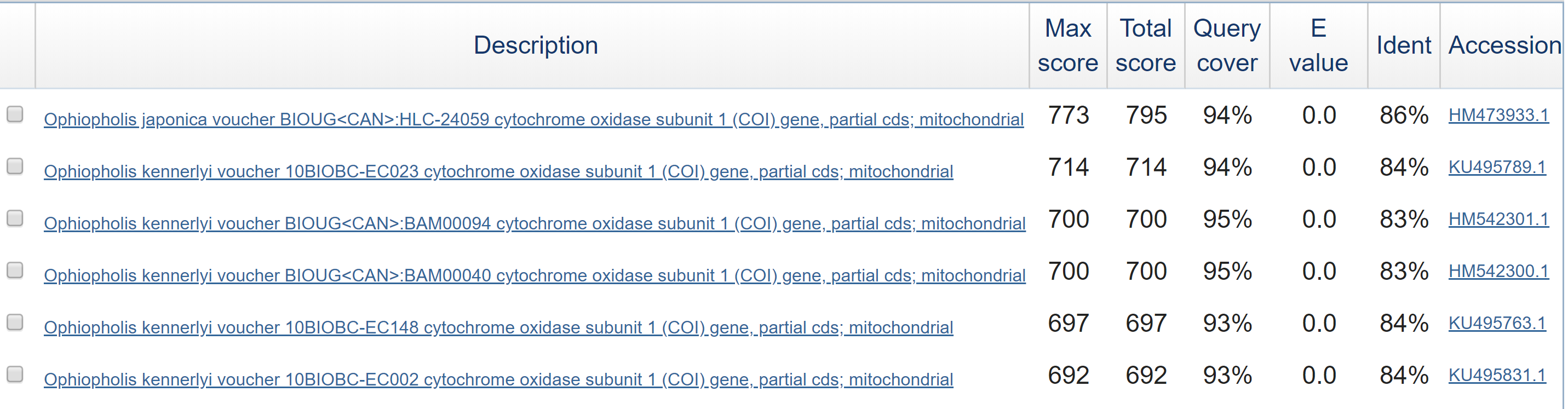
Рисунок 3. blastn с параметрами по умолчанию (последовательность с хроматограммы, исключен вид). Первая находка та же самая, потом другие, всего 23. На самом деле там почти все находки одного вида Ophiopholis kennerlyi (но полученные в разных экспериментах). У всех находок, кроме последней, стоит Е=0. Удивляюсь, вспоминаю, что Е зависит от параметров поиска, поэтому сравнивать с предыдущей картинкой нельзя. Также Е зависит от банка, по которому ищем, а тут наиболее похожие последовательности (вида Ophiopholis aculeata) исключены, поэтому ожидаемое количество находок с лучшим соответствием действительно ноль.
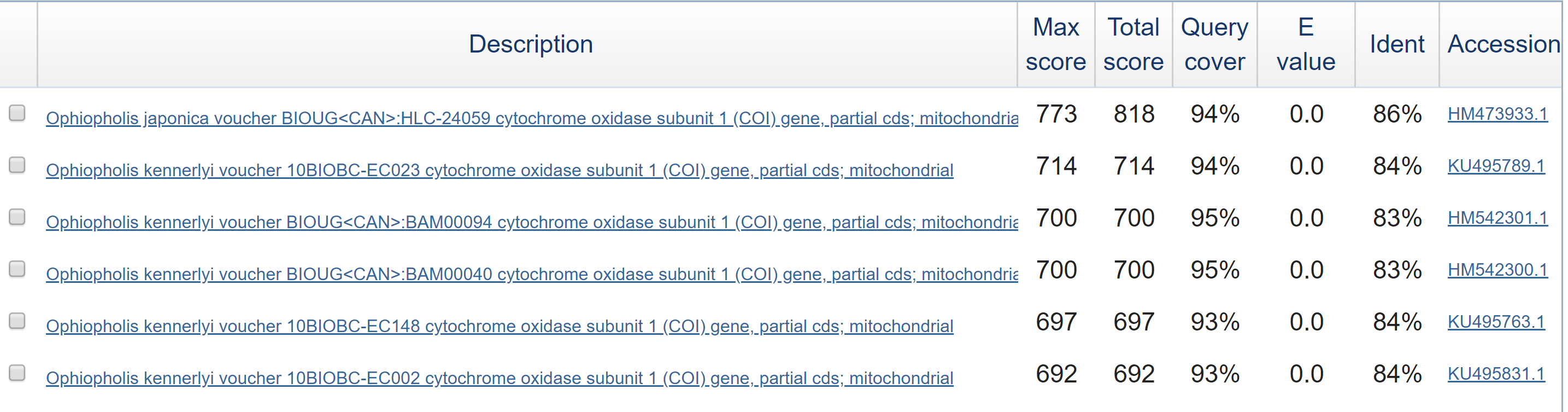
Рисунок 4. Это картинка из другого поиска, но начало выглядит точно так же :) Находок больше (32), лучшая находка та же самая. Точнее, первая половина списка находок та же самая, а потом чуть-чуть меняется порядок. В конце почему-тодобавились две последовательности с покрытием 2% и 1% (у предыдущей перед ними находки 69%), это единственные последовательности какого-то другого белка среди находок) Там длина совпадения 11 и 17 нуклеотидов - многовато для случайного совпадения (?), но маловато (?) для какого-то похожего функционального участка. Зато видна чувствительность алгоритма: при стандартных настройках этих находок не было.
Для дальнейших сравнений работы алгоритма была выбрана последовательность 18S рРНК из митохондриального генома (практикум семь, Яблоня домашняя, координаты рРНК 275318..277180). Последовательность была скачана в формате fasta (я нашла ее по GeneID, можно было вместо этого вырезать из митохондриального генома по координатам).
Blastn со стандартными настройками нашел очень много похожих находок (много сортов яблонь), поэтому для трёх дальнейших поисков я исключила из поиска род Malus, а также ограничила поиск цветковыми растениями, чтобы не тратить время. В каждом случае нашлось 100 находок (на самом деле больше, но выводятся не более 100 лучших). Топ-список находок у алгоритмов был разный, но никакой закономерности не наблюдалось. Видимо, это связано с консервативностью рибосомальных РНК и 18S в частности. Я решила попробовать поискать гомологи среди Архей. Находок всё еще больше 100 при всех трех алгоритмах, первые находки везде очень хорошие. Предсказуемо, что все находки - рРНК 16S.
У megablast находок 125, у blastn со стандартными параметрами - 203, у blastn повышенной чувствительности (длина слов меньше и match/mismatch изменены с 3/-2 на 1/-1) находок 253. Т.е. видно, что megablast настроен на поиск очень похожих последовательностей, поэтому (несмотря на консервативность рРНК) с ограничением на другой домен находит меньше, чем blastn, а blastn в свою очередь меньше, чем он же с более чувствительными параметрами. При этом в этой линии из трех алгоритмов не только увеличивается число находок, но и повышается их качество: в этом ряду значения E-value сотой находки соответственно 1e-76, 3e-96, 4e-120.
[Аннотация генома - описание (предсказание) его функциональных и структурных характеристик. Т.е. в неаннотиованном геноме не предсказаны и не описаны белки.]
Для поиска с помощью локального BLAST (на kodomo, не на локальном компьютере), из сборки генома Amoeboaphelidium protococcarum была создана база данных. Т.к. нужно найти гомологи белка в нуклеотидной базе данных, был выбран алгоритм tblastn. Последовательности белков, гомлоги которых нужно найти в геноме, были скачаны в формате fasta. Это можно было сделать через сайт UniProt (и заодно почитать про эти белки) или по идентификатору с помощью seqret.
Поиск запсукался такой командой (чтобы было удобнее анализировать, я перенаправила результаты в файл):
seqret sw:hsp71_yeast hsp71_yeast.fasta tblastn -query hsp71_yeast.fasta -db X5.fasta > hsp71_yeast_blast.txt
Таблица 1. Поиск гомологов трех белков.
| белок | всего находок | положение в геноме лучших находок | вес в битах лучшей находки | Е-value лучшей находки | Positives лучшей находки | Вывод |
| HSP71_YEAST | 16 | scaffold-199 | 920 | 0.0 | 90% | Первые три находки (в составе scaffold-199, scaffold-96, scaffold-423 соответственно) имеют битовый вес > 700 и высокое покрытие искомой последовательности, а также длинные консервативные участки. Можно утверждать, что эти три находки - гомологи данного белка. |
| TBB_NEUCR | 5 | unplaced-665 | 742 | 0.0 | 88% | В начале выравнивания много несовпадающих позиций и один довольно длинный разрыв, но после него идёт длинный консервативный участок длиной более 350 а.о., что составляет большую часть длины данного белка. Белки гомологичны. |
| TERT_SCHPO | 4 | scaffold-17 | 108 | 1e-23 | 47% | Низкий вес выравнивания, высокое число гэпов и отсутствие сколько-ниудь длинных консервативных участков не позволяют предположить, что лучшая находка гомологична данному белку. |
Самый длинный из имеющихся скэффолдов scaffold-17 имеет длину более 2 миллионов нуклеотидов. Сначала я пробовала найти белок в scaffold-420 и scaffold-242, но большинство нашедшихся белков были гипотетическими, то есть для них не была предсказана функция. Попытка найти белок в scaffold-223 (длиной 76170 нуклеотидов) оказалось более успешной.
Последовательность выбранного скэффолда вырезалась в отдельный файл. Поиск производился на сервере NCBI алгоритмом blastx (алгоритм ищет в белковой базе данных, на вход принимает нуклеотидную последовательность). Искались похожие последовательности в базе данных "Reference proteins", ограничение по организму - Fungi(taxid:4751). Вот лучшие находки:
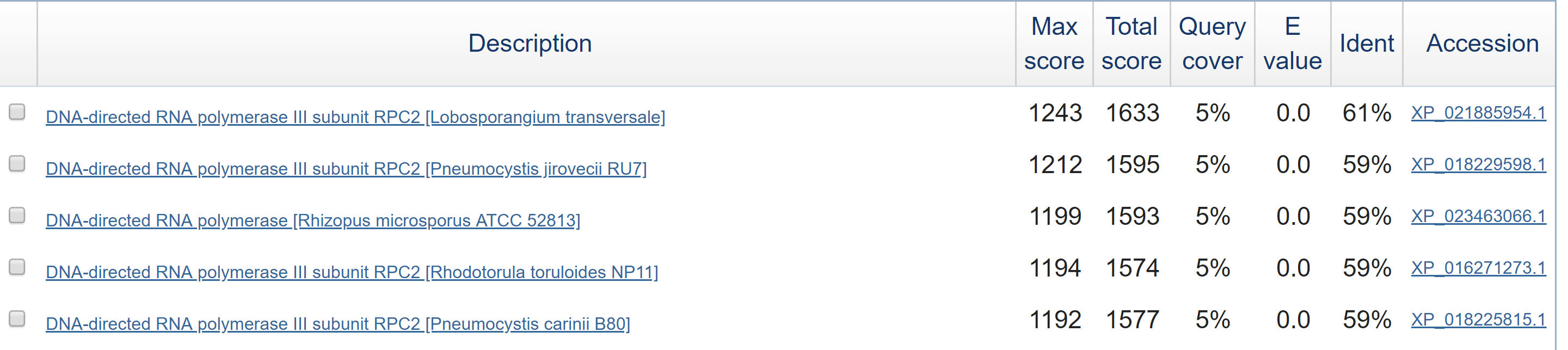
Рисунок 5. Поиск белка в scaffold-223.
Вся верхняя часть списка находок относится к белкам DNA-directed RNA polymerase III subunit RPC2 разных организмов. В нижней части списка встречаются также записи о beta and beta-prime subunits of RNA-polymerase, у них процент идентичности похожий, но покрытие заметно меньше, видимо, это другие субъединицы полимеразы, имеющие гомлогичные участки с субъединицей из лучших находок. Можно утверждать, что в scaffold-223 содержится ген субъединицы RPC2 ДНК-зависимой РНК-полимеразы третьей. Не очень высокий процент идентичности (около 60%) может быть связан с тем, что лучшие находки - не очень близкие родственники Amoeboaphelidium (как мне удалось понять по ссылке 'Taxonimy'), мне также показалось немного странным, что не нашлось гомологов в более близких родственниках. Возможно, это связано с тем, что геномов более близких родственников нет в базе Reference proteins, которой был ограничен поиск.
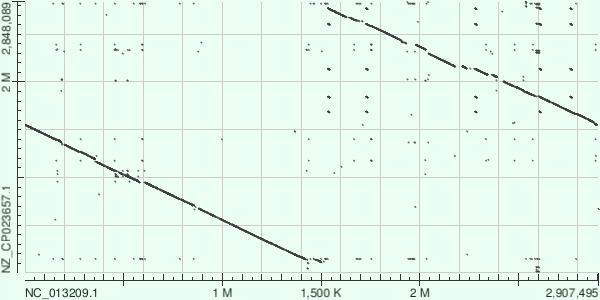
Для выполнения этого задания был выбран род бактерий Acetobacter. Карты локального сходства были построены на сайте NCBI алгоритмом megablast (т.к. предполагается, что геномы бактерий одного рода очень похожи).
| | |
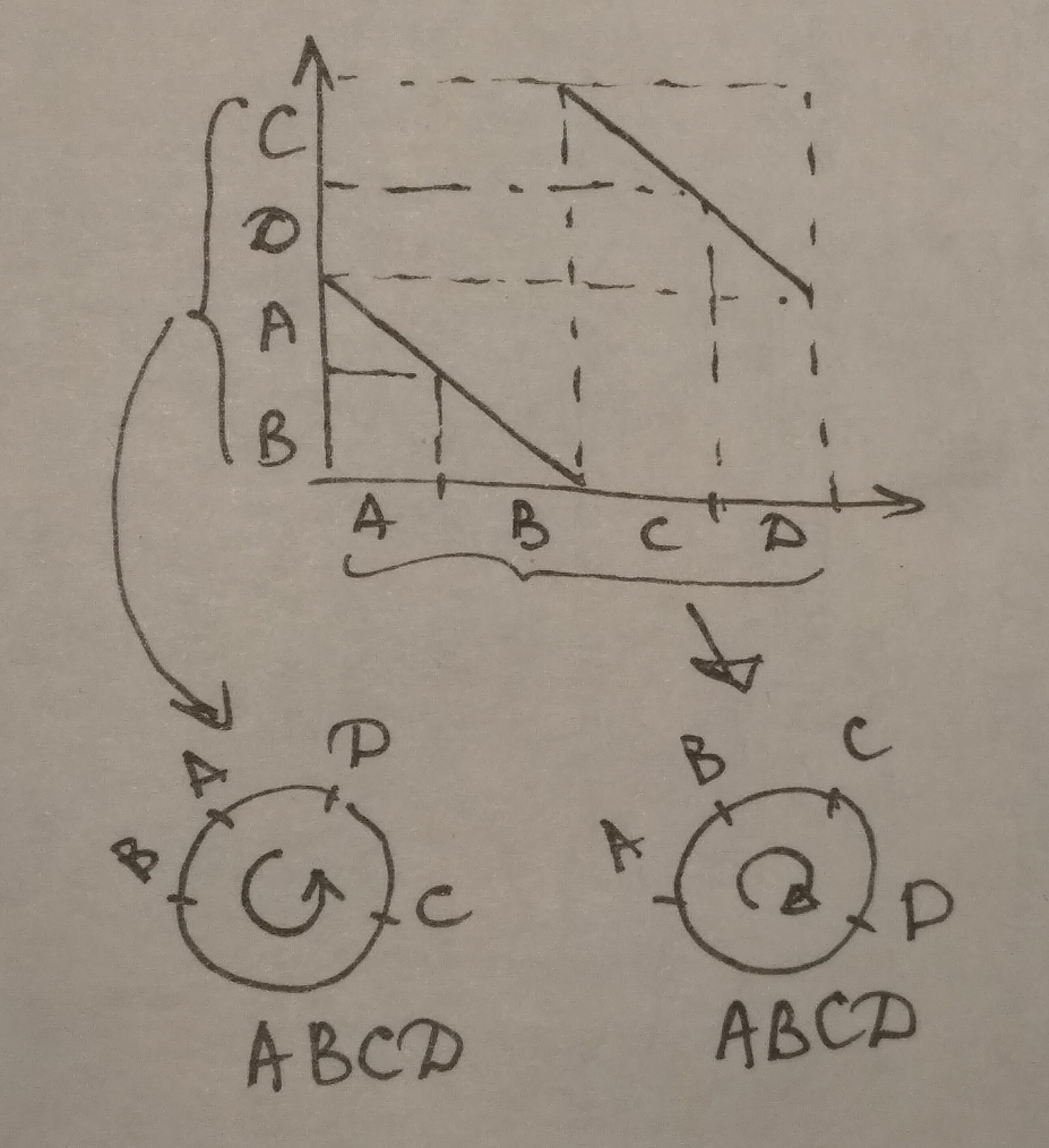
| Рисунок 6. Карта локального сходства для Acetobacter pasteurianus IFO 3283-01 (по горизонтальной оси, идентификатор NC_013209.1) и Acetobacter pomorum (по вертикальной оси, идентификатор генома NZ_CP023657.1). Карта построена при стандартных параметрах алгоритма megablast и слабо меняется при изменении параметров. | Рисунок 7. - графическое объяснение того, что на рисунке 6 геномной перестройки нет. |
Между этими двумя геномами нет транслокации (точнее есть, но они точечные). На рисунке 7 последовательность, отложенная по горизонтальной оси, условно разделена на две части (по тому месту, где я сначала предположила разрыв). Далее каждая из частей обозначена двумя буквами, однозначно задающими направление (условно начало и конец). Далее по карте сходства составляем вторую последовательность. Получается, что она отличается от первой на циклическую перестановку и смену направления (что для кольцевой хромомсомы не имеет значения).
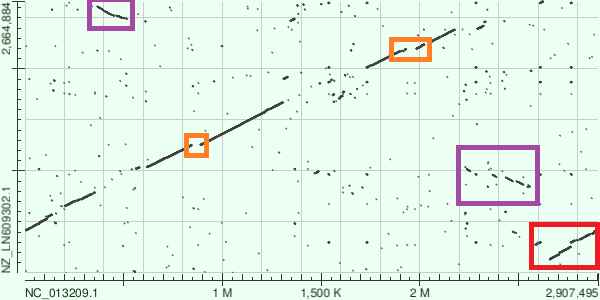
Рисунок 8. Карта локального сходства для того же Acetobacter pasteurianus IFO 3283-01 (по горизонтальной оси, идентификатор NC_013209.1) и Acetobacter ghanensis (вертикальная ось, идентификатор NZ_LN609302.1). Карта построена алгоритмом megablast, но уменьшены штрафы за гэпы (по умолчанию они линейно зависят от длины гэпа). Красным цветом выделена транслокация, фиолетовым - инверсии с транслокацией, оранжевым - делеции в геноме, отложенном по вертикальной оси.
Вернуться на страницу семестра
© potapenko 2017-2018