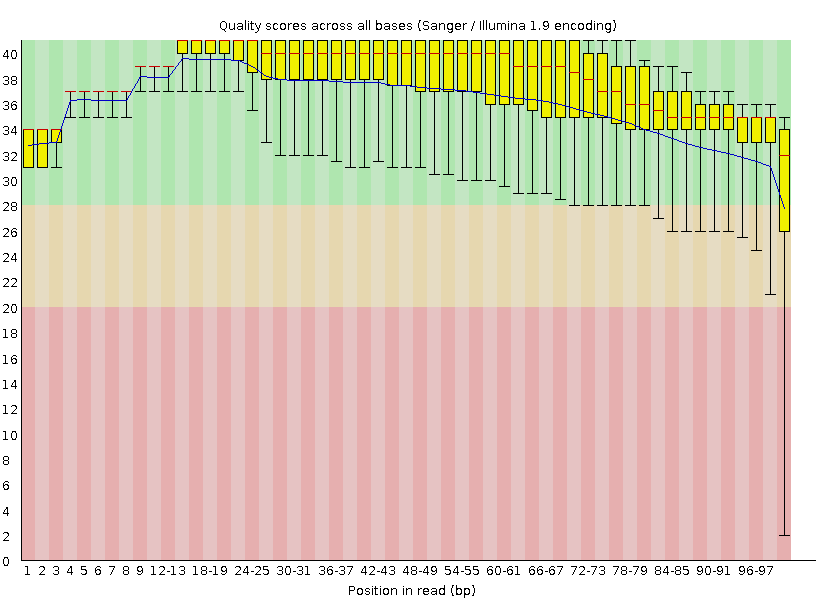
График поделён на три области: зелёную, жёлтую и красную,
соответствующие качеству ридов.
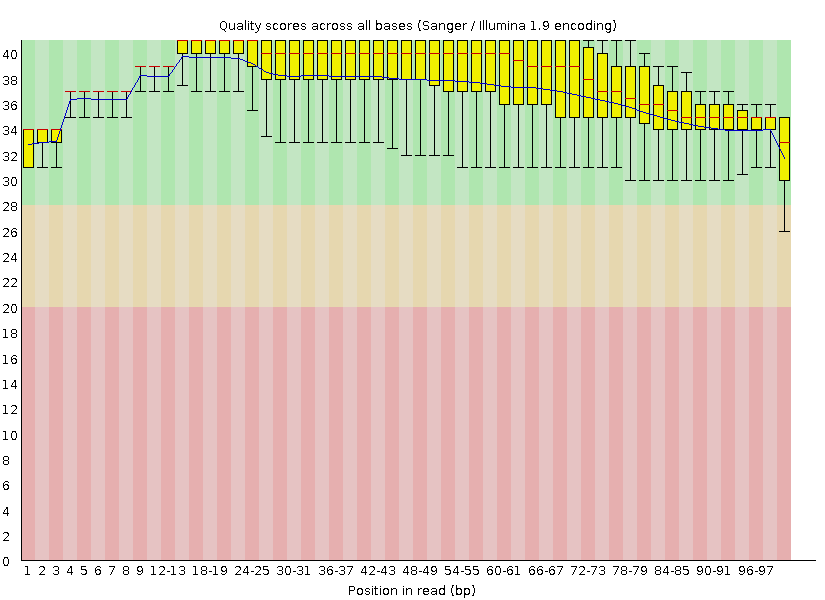
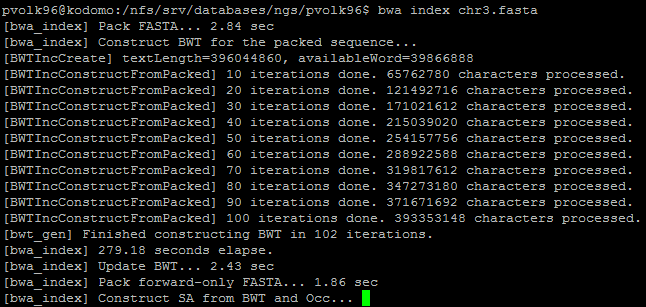


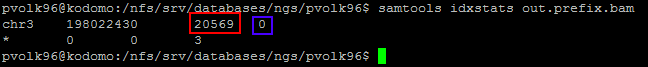

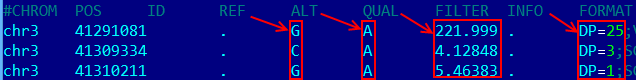
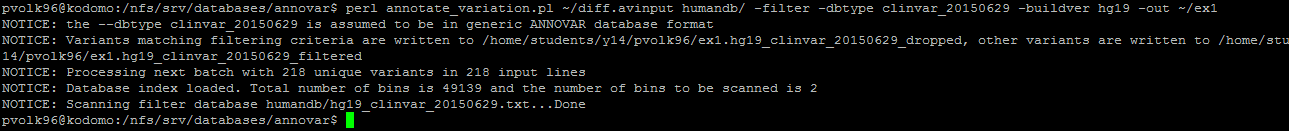
Таблица 1. Команды, которые были использованы в практикуме. | |
Команда |
Результат |
| fastqc chr3.fastq | анализ качества ридов |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr3.fastq trailing.fastq TRAILING:20 | Очистка чтения: удаляет с конца рида нуклеотиды с качеством ниже 20 |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 trailing.fastq a.fastq MINLEN:50 | Очистка чтения: удалены риды с длиной менее 50 нуклеотидов. |
| bwa index chr3.fasta | Индексирует референс |
| bwa mem chr3.fasta a.fastq > BW.sam | Картирование ридов на хромосому 3: выравнивание строит чтений с референсной последовательностью |
| samtools view BW.sam -b -o BW.bam | Перевод файла с выравниванием в бинарный формат (.bam) |
| samtools sort BW.bam out.prefix | сортирует выравнивание чтений с референсом. |
| samtools idxstats out.prefix.bam | Определяет количество картированных чтений |
| samtools mpileup -uf chr3.fasta out.prefix.bam > polymorfism.bcf | Создание файла с полиморфизмами |
| bcftools call -cv polymorfism.bcf > dif.vcf | Перевод .bcf файла в формат .vcf. |
| perl convert2annovar.pl -format vcf4 ~/dif.vcf > ~/diff.avinput | Перевод файла .vcf в файл .avinput, с которым может работать Annovar. |
| perl annotate_variation.pl -filter -out ~/rs -build hg19 -dbtype snp138 ~/diff.avinput humandb/ | Аннотация файла со списком SNP по БД refgene. |
| perl annotate_variation.pl -out ~/ref.out -build hg19 ~/diff.avinput humandb/ | Аннотирует файл со списком SNP по базе данных dbsnp. |
| perl annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out ~/1000.out ~/diff.avinput humandb/ | Аннотирует файл со списком SNP по базе данных 1000genomes. |
| perl annotate_variation.pl -regionanno -build hg19 -out ~/gwas.out -dbtype gwasCatalog ~/diff.avinput humandb/ | Аннотация файла со списком SNP по базе данных GWAS. |
| perl annotate_variation.pl ~/diff.avinput humandb/ -filter -dbtype clinvar_20150629 -buildver hg19 -out ~/clinvar.out | Аннотация файла со списком SNP по базе данных Clinvar |