








Парное выравнивание подразумевает выравнивание только двух последовательностей. В отличие от множественного, оно гораздо менее надежно, поэтому в этом задании используются несколько методов построения и проверки парного выравнивания.
1. Получение парного выравнивания из множественного.
Было взято множественное выравнивание, из которого были удалены все последовательности, кроме двух последних. Пустые колонки были удалены.
• Получившееся выравнивание можно скачать здесь: pairwise.mfa
2. Глобальное выравнивание.
Глобальное выравнивание - это выравнивание последовательностей по всей длине. Оно осуществляется по алгоритму Нидлмана-Вунша. Были взяты те же две последовательности, что и в пункте 1. Выравнивание выполнялось с помощью программы needle из пакета EMBOSS.
• Глобальное выравнивание в формате .fasta можно скачать здесь: global.fasta
3. Локальное выравнивание.
Локальное выравнивание, в отличие от глобального, выбирает гомологичные участки обоих поледовательностей и строит выравнивание между ними. Локальное выравнивание осуществляется по алгоритму Смита-Ватермана. Выравнивание выполнялось с помощью программы water из пакета EMBOSS.
• Локальное выравнивание в формате .fasta можно скачать здесь: local.fasta
4. Ложное выравнивание.
Было построено парное выравнивания последовательностей двух заведомо негомологичных белков: 2JG8_A (узнающая единица комплекса C1, участвующего в апоптозе) и взятого из другого выравнивания белка 4CJM (фактора роста фибробласта). Для этих белков было построено "выравнивание" программами needle и water.
• Ложные выравнивания (глобальное и локальное) в формате .fasta можно скачать здесь: globalfake.fasta, localfake.fasta
Было посчитано количество совпадающих, схожих аминокислот, гэпов и открытий гэпов во всех полученных последовательностях. Это достаточно просто посчитать, если при запуске needle и water сохранять не в .fasta, а в их формате по умолчанию. В этом формате совпадающие аминокислоты отмечены знаком "|", схожие - ":", а гэпы отмечены чертой "-". Для выравнивания, полученного в JalView в пункте 1, подобный формат можно получить через Calculate -> Pairwise alignments, правда, в таком случае схожие аминокислоты будут отмечены "." Пример такого формата: глобальное выравнивание
Табл.1. Параметры полученных в пунктах 1-4 выравниваний.
|
Выравнивание |
Длина выравнивания |
Число совпадений |
Процент совпадений |
Число схожих |
Процент схожих |
Число гэпов |
Процент гэпов |
Число открытий гэпов |
|
1 (из множественного) |
137 |
45 |
32.8% |
30 |
21.9% |
8 |
5.8% |
6 |
|
2 (глобальное) |
159 |
50 |
31.4% |
15 |
9.4% |
55 |
34.6% |
7 |
|
3 (локальное) |
151 |
50 |
33.1% |
15 |
9.9% |
50 |
33.1% |
7 |
|
4 (ложное глобальное) |
209 |
19 |
9.1% |
15 |
7.2% |
155 |
74.2% |
6 |
|
5 (ложное локальное) |
42 |
12 |
28.6% |
9 |
21.4% |
6 |
14.3% |
2 |
Видно, что первое выравнивание лучше (больший процент схожих аминокислот и меньше гэпов), чем второе или третье, которые примерно одинаковы по качеству. Ложное глобальное выравнивание показало огромное количество гэпов и малый процент совпадения, а вот локальное ложное построило небольшой, но хорошо совпадающий участок.
5. Сравнивание двух выравниваний.
На этом этапе я сравнила последовательность, полученную из множественного выравнивания, с полученной программой needle. Второе выравнивание было добавлено к первому в JalView, и, двигая пары друг относительно друга, я попыталась добиться наибольшего совпадения колонок. Результат можно увидеть в готовом проекте (ссылка в начале страницы) или скачать fasta-файл
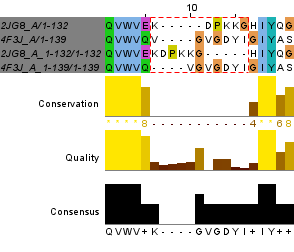
Рис.1. Участок с малым совпадением выравниваний (выделен), окруженный совпадающими участками. Координаты участка с несовпадением: 125-135.
7. Проверка правильности выравниваний
В проекте JalView в окне со сравнением выравниваний были добавлены 4 строки с аннотациями, по 2 на каждое из двух выравниваний. Знаком "+" отмечены колонки, в которых можно предположить гомологию последовательностей. В первом выравнивании гомология прослеживалась из консервативных колонок множественного выравнивания, а во втором - только из предположения о гомологии длинных совпадающих участков последовательностей. Поэтому в первом выравнивании могут быть отмечены плюсом стоящие отдельно колонки, которые показали консервативность во множественном выравнивании, а во втором - нет.
Знаком "s" отмечены колонки, в которых выравнивание совпадает с совмещением атомов в наложенных трехмерных структурах белков. Для обоих выравниваний были получены скрипты RasMol программой SupCheck, затем скрипт был запущен в RasMol на совмещенных PDB-структурах белков. Работа скрипта показывала, как атомы из данной колонке расположены на совмещенных структурах.
Для обоих выравниваний затем были посчитаны ошибки 1 и 2 рода.
Выравнивание, полученное из множественного:
- Ошибки 1 рода (атомы в колонке, отмеченной "+", не совмещаются в структурах) - 1
- Ошибки 2 рода (атомы, совмещающиеся на структурах, но соответствующие буквы либо не находятся в одной колонке, либо в одной колонке, но колонка не отмечена "+") - 46
Глобальное выравнивание:
- Ошибки 1 рода - 0
- Ошибки 2 рода - 83
В целом, второе выравнивание было хуже, там было 2 довольно длинных участка, где два белка хорошо совмещались, но в выравнивании эти атомы были разнесены.