








Программа getorf
С помощью команды entret embl:D89965 была получена запись D89965 банка EMBL. В записи содержится последовательность гена белка крысы (Rattus norvegicus), ответственного за передачу сигналов с помощью серотонин-связывающего рецептора.
Команда getorf выдает все возможные рамки считывания (на основании старт- и стоп-кодонов) данной последовательности белка или нуклеиновой кислоты. Мне нужно было получить рамки, определенные с помощью стандартного генетического кода, имеющие длину не менее 30 а.о., начинающиеся со старт-кодона и заканчивающиеся стоп-кодоном. Для этого я выполнила getorf с параметрами:
getorf d89965.entret -table 0 -minsize 30 -find 3
Вообще минимальный размер (minsize) по умолчанию уже стоит 30, а таблица генетического кода (-table) указана стандартная [0]. Поиск (-find) по умолчанию производится на транслированные последовательности от стоп-кодона до другого стоп-кодона, поэтому нужно указать другой параметр, выдающий именно нуклеотидные последовательности между стартом и стопом. В итоге командную строку можно записать проще:
getorf d89965.entret -find 3 -auto
Кодирующую последовательность из записи embl можно получить командой seqret -sask с параметрами: начало - 163 позиция, конец - 435 позиция (рамки CDS, указанные в записи embl). Получается последовательность:
>D89965 D89965.1 Rattus norvegicus mRNA for RSS, complete cds. atggccctgatgcatttccagtttacgttcaaacagttcgaacagcgtaaaagcatccgc agtaccgcccgcaaagcccgcgatgactttgtcgttgtacagacggcggacctttttcac gttgcctttcattacggtattgcccaacgtggcctgaccatcaccagcgatgaccacatg gccgttacggcgtacgcttactatagttgtcacgagctgaccccttggttacgaatacag agtacaaaccccgtacaaaagtacggggcataa
Из возможных рамок считывания, выданных getorf, этому соответствует:
>D89965_5 [163 - 432] Rattus norvegicus mRNA for RSS, complete cds. atggccctgatgcatttccagtttacgttcaaacagttcgaacagcgtaaaagcatccgc agtaccgcccgcaaagcccgcgatgactttgtcgttgtacagacggcggacctttttcac gttgcctttcattacggtattgcccaacgtggcctgaccatcaccagcgatgaccacatg gccgttacggcgtacgcttactatagttgtcacgagctgaccccttggttacgaatacag agtacaaaccccgtacaaaagtacggggca
Последовательности идентичны, кроме последнего триплета (стоп-кодон taa).
Файлы-списки
В этом задании нужно было найти последовательности всех алкогольдегидрогеназ (ADH) из базы Swiss-Prot, принадлежащих данным 7 организмам. Во-первых, получить все алкогольдегидрогеназы из Swiss-prot можно командой seqret sw:adh* adh.fasta, где seqret проведет поиск всех записей с AC, начинающимся с "adh", и запишет все их последовательности в файл adh.fasta. Но это не обязательно. Чтобы собственно выполнить задачу, сначала я получила универсальные адреса (USA) этих алкогольдегидрогеназ. Для этого я воспользовалась командой infoseq, которая выдает на выбор какую-либо краткую информацию о последовательности. Команда infoseq sw:adh* -outfile list -only -usa запишет USA всех алкогольдегидрогеназ из Swissprot в файл list. Затем нужно было выбрать из них адреса последовательностей, принадлежащим сделующим организмам: Человек (HUMAN), кряква (ANAPL, Anas platyrhynchos), плодовая муха (SCACA, Scaptomyza crassifemur), три дрозофилы (DROFL, DROBU и DROMM) и грызун с очаровательным названием Knox Jones's pocket gopher (GEOKN). Адреса последовательностей были отобраны grep в новый файл-список. Для этого я произвела команду grep -f species list > list_less , где -f - опция grep, говорящая ему принять список слов для поиска из файла species, где каждое слово (идентификатор организма) записано на новой строке. Он затем был использован в команде seqret @list_less adh_mine.fasta, где @ - особенность EMBOSS, говорящая программе, что на вход подается файл-список.
- Файл со списком нужных последовательностей: adh_mine.fasta
Случайная модель для оценки достоверности выравнивания
Как оценить достоверность выравнивания? Можно ли из него понять, что гены гомологичны? Очевидно, что для этого можно сравнить вес нужного выравнивания с весами выравниваний одной из последовательностей с последовательностями, полученными случайным образом. Я взяла последовательности двух алькогольдегидрогеназ, принадлежащих крякве (Anas platyrhynchos) и плодовой мухе (Scaptomyza crassifemur). Идентификаторы, соответственно, ADH1_ANAPL и ADH_SCACA. Выравнивание:
ADH1_ANAPL 8 VFGLGGVGLSVIMGCKAAGASRIIAVDINSDKFAKAKELGATDCINPKDH 57 |.||||:||........:|...::.:| .:...|...||.| :|| ADH_SCACA 11 VAGLGGIGLETSREIVKSGPKNLVILD-RAPNPAAIAELQA---LNP--- 53 ADH1_ANAPL 58 KEPIHKVLIGMTGYGVDY-SFEVIGRIETMVAAL 90 ||.:....|.|.. ..|.:..::|:.|.| ADH_SCACA 54 -----KVTVSFYLYDVTVPQSETVKLLKTIFAKL 82
Затем первая последовательность (из кряквы) была случайно перемешана 100 раз командой shuffleseq:
shuffleseq sw:ADH1_ANAPL ADH_shuffled.fasta -shuffle 100
Были построены 100 попарных выравниваний командой water последовательности ADH_SCACA с перемешанными последовательностями. Был получен файл со 100 выравниваниями в формате water. В этом формате также указывается вес (Score) каждого выравнивания. Поэтому список весов был получен командой grep 'Score' shuffled.fasta > Score. По этому файлу была построена диаграмма в Excel.
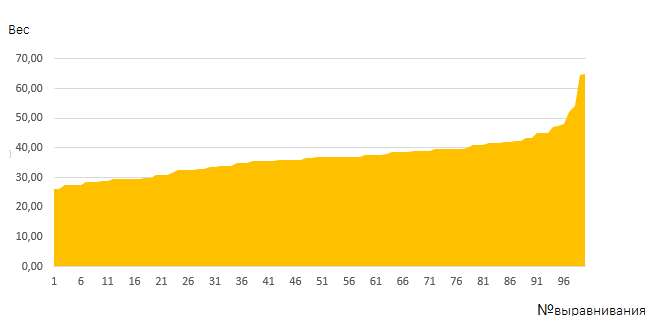
Распределение весов в построенных выравниваниях (выравнивания были сортированы по возрастанию весов).
Вес выравнивания наших алкогольдегидрогеназ составил 53.5. Три выравнивания со случайными последовательностями превзошли по весу выравнивание предположительно гомологичных. То есть хотя вес нужного выравнивания гораздо выше большинства других весов, однозначно судить о гомологичности по весу в общем-то нельзя.