Комплексы ДНК-белок¶
1. Предсказание вторичной структуры заданной тРНК¶
Нужно было использовать различные алгоритмы дляпредсказания вторичной структуры РНК исравнить их с реальной вторичной структурой РНК. Сначала воспользовались программой einverted:

Затем вторичная структура РНК была предсказана по алгоритму Зукера. У исходной тРНК (2cv0) алгоритм Зукера предсказал 21 пару азотистых оснований.
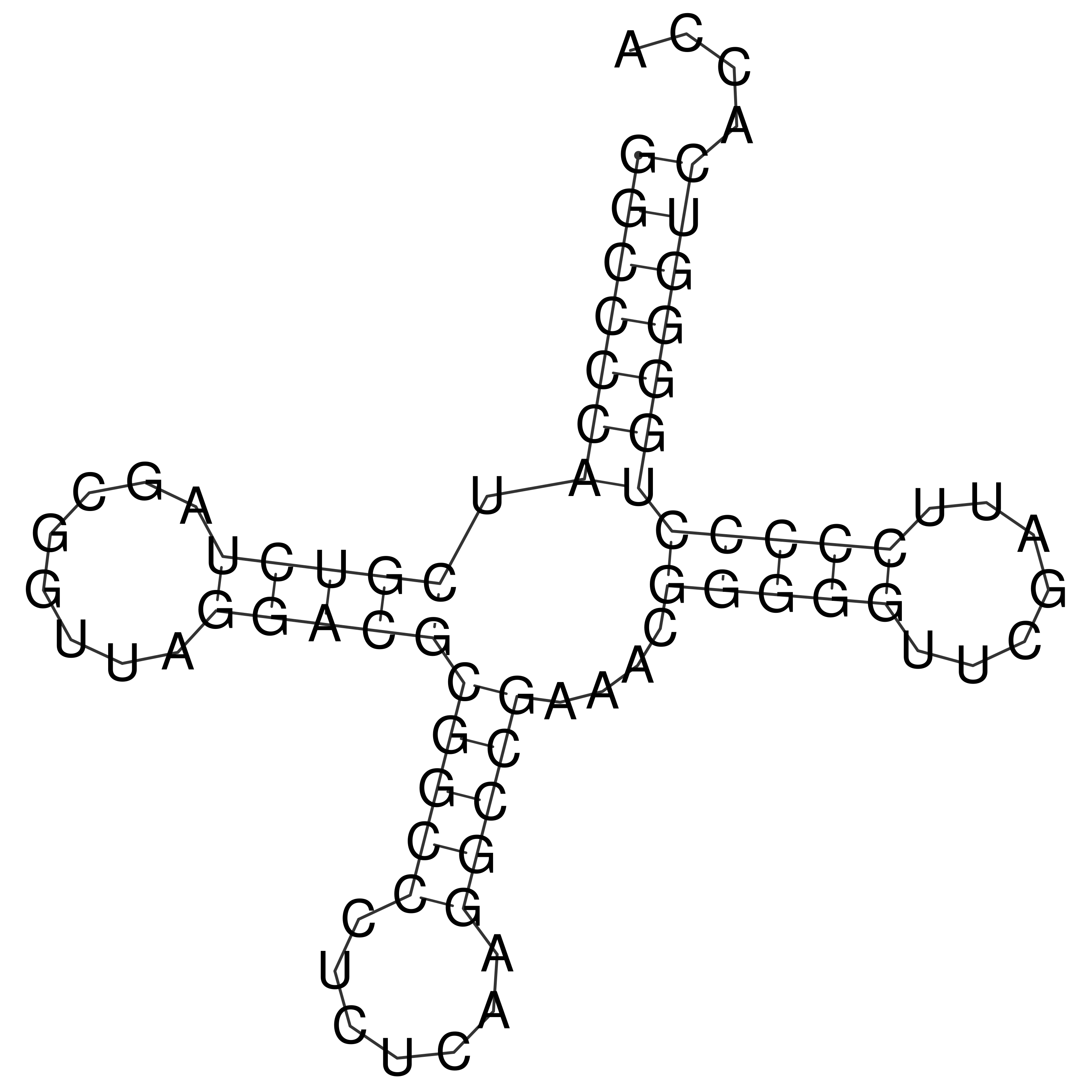
Реальная и предсказанная вторичная структура тРНК из файла 2cv0.pdb¶
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5' -1-7- 3' 5'-66-72-3' |
предсказано 5 пар из 7 реальных | Предсказаны Все пары |
| D-стебель | 5'-49-53-3' 5'-61-65-3' |
предсказано 0 пар | Предсказаны все пары |
| T-стебель | 5'-39-44-3' 5'-26-31-3' |
предсказано 0 пар | Предсказаны все пары |
| Антикодоновый стебель | 5'-10-15-3' 5'-20-25-3' |
предсказано 0 пар | Предсказаны все пары |
| Общее число канонических пар нуклеотидов | 22 | 5 | 22 |
По таблице видно, что алгоритм Зукера точнее предсказывает структуру РНК.
2. Поиск ДНК-белковых контактов в заданной структуре¶
Для нахождения контактов ДНК с белком был написан скрипт, который можно посмотреть здесь: Script
Контакты атомов белка с | Полярные | Неполярные | Всего |
остатками 2'-дезоксирибозы | 7 | 47 | 54 |
остатками фосфорной кислоты | 25 | 29 | 54 |
остатками азотистых оснований со стороны большой бороздки | 21 | 0 | 21 |
остатками азотистых оснований со стороны малой бороздки | 13 | 6 | 19 |
Результат выполнения алгоритма nucplot для 1dfm:

 Аминокислотный остаток с наибольшим числом контактов: ASP 84 (4 контакта)
Аминокислотный остаток с наибольшим числом контактов: ASP 84 (4 контакта)
 На мой взгляд, ASN 140 - наиболее важный для распознавания последовательностей, т.к. образует много контактов, причём затрагивает обе цепи
На мой взгляд, ASN 140 - наиболее важный для распознавания последовательностей, т.к. образует много контактов, причём затрагивает обе цепи
