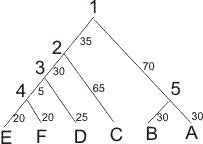
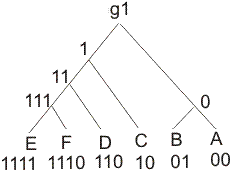
6 00 0.0000 0.5034 1.2466 1.3956 1.1944 1.4467 01 0.5034 0.0000 1.4293 1.5609 1.3956 1.6031 10 1.2466 1.4293 0.0000 0.8317 0.8798 0.9492 110 1.3956 1.5609 0.8317 0.0000 0.4136 0.3704 1110 1.1944 1.3956 0.8798 0.4136 0.0000 0.2945 1111 1.4467 1.6031 0.9492 0.3704 0.2945 0.0000
На основе этого выравнивания были расчитаны расстояния по Джуксу - Кантору с помощью программы ednadist из пакета EMBOSS. При этом в качестве параметра "Transition/transversion ratio" мы использовали "1.0", поскольку в нашей эволюционной модели транзиции и трансверсии не различались.