 Terms
Terms
|
 Other results
Other results
|
 Home page
Home page
|
| |
|
Отчет по практису 8
-
- Данная структура имеет pdb код -
1O0B
- Это структура тРНК(GLUTAMINYL TRNA) комплекса вместе с белком синтазой(Лигазой) - GLUTAMINYL-TRNA SYNTHETASE(LIGASE)
тРНК здесь представленна одной цепью (B) длинной 75 нуклеотидов
>-
Последовательность ДНК выглядит так:
UGGGGUAUCGCCAAGCGGUAAGGCACCGGAUUCUGAUUCCGGCAUUCCGAGGUUCGAAUCCUCGUACCCCAGCCA
Последовательность начинается с 902 номера и заканчиваеться 976 исключая 917 номер.
-
По выдаче программой find_pair
U GGGGUA
UC
GCC
AAGCGGUAA
GGC
ACCGGAUU
CUG
AUUCCGGC
AUUC
CGAGG
UUCGAAU
CCUCG
UACCCC
AGCCA
Соответственно если разметить аналогично pdb структуру(справа) и вручную сотставленая модель(слева):
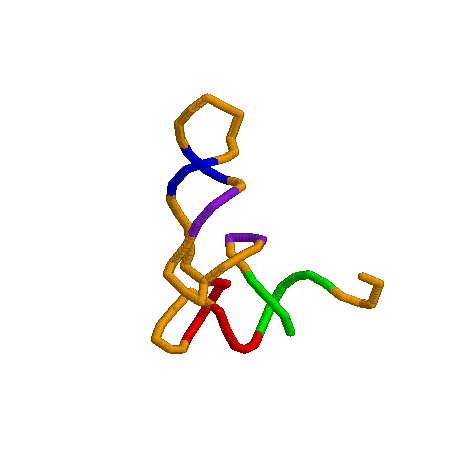
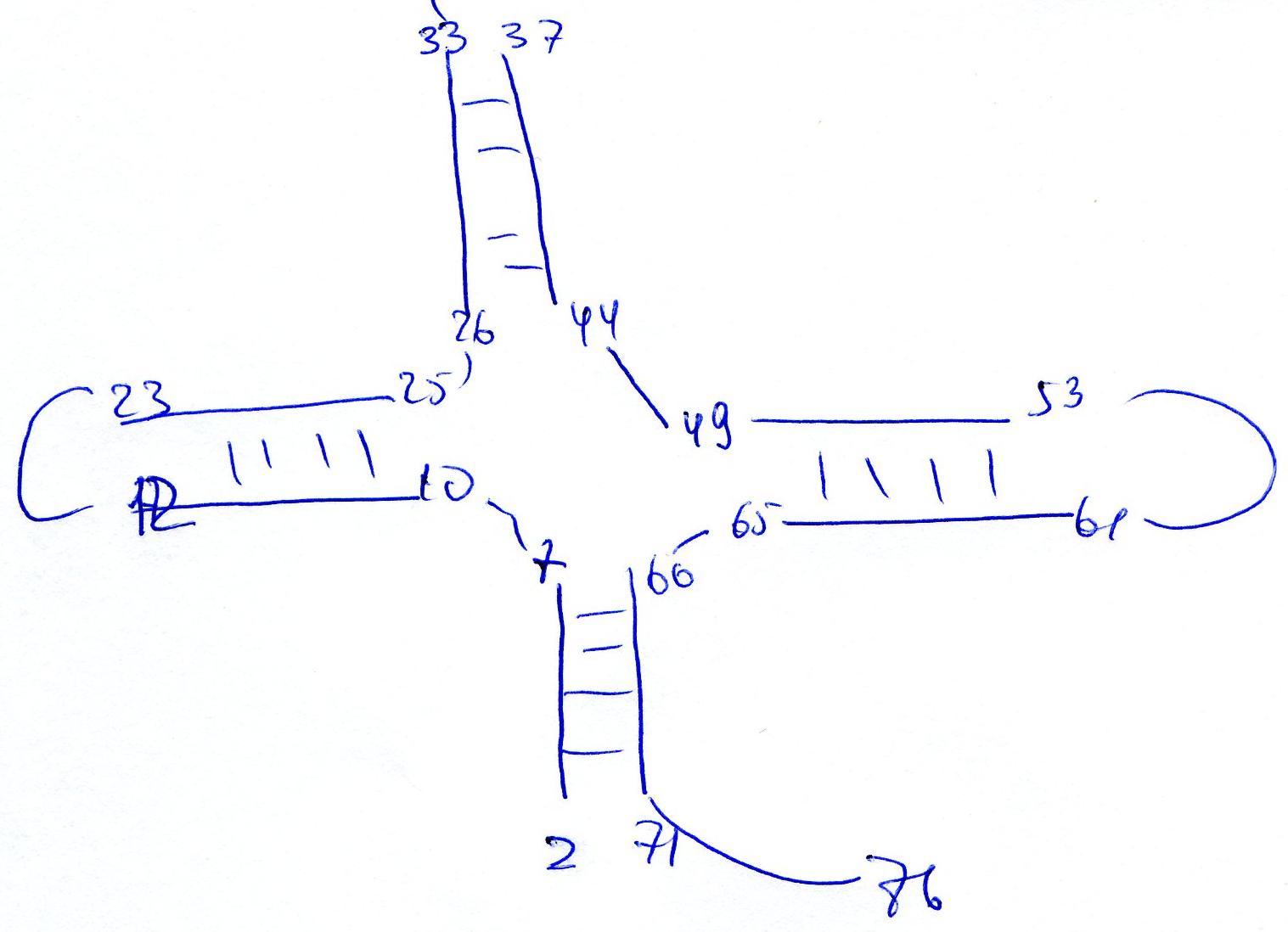
Скрипт создающий размоловское изображение tRNA_Script_2
-
-
Пример внеспирального стекинг взаимодействия:
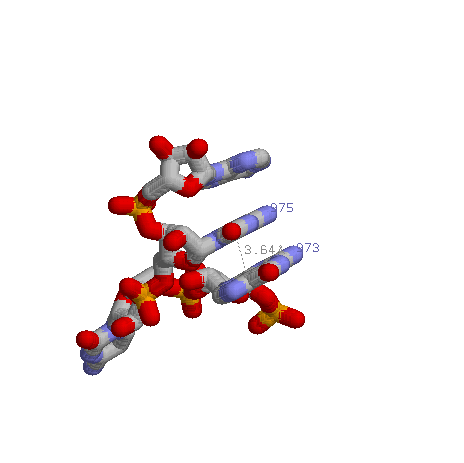
-
Пример водородных связей между основаниями, не сводящийся к
Уотсон-Криковскому спариванию комплементарных оснований;
Картинка приведена только для одной такой связи полный список представлен в файле
который выдает analyze :
"##### 8 non-Watson-Crick base-pairs, and 5 helices (1 isolated bp)"
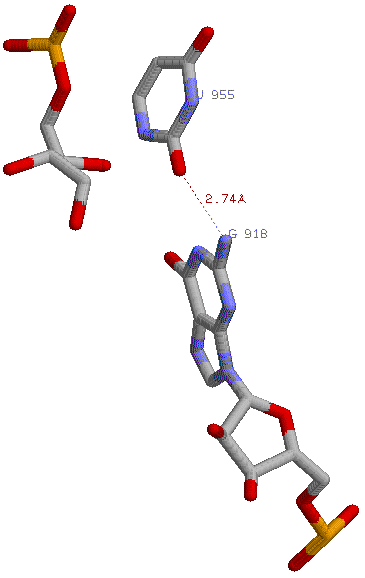
-
В файле выдаваемом программой analyze для спиралей указываеться форма. Плюс к этому как доказательство можно привести вид с торца:
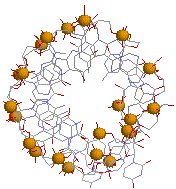
По отверстию внутри спирали можно утверждать, что это А форма.
-
Исправления :pdb файл действительно отличался видно предыдущий скачался с ошибками, но последовательности одинаковы, а значит и результаты последних двух заданий - неизменны.
UGGGGUAUCGCCAAGCGGUAAGGCACCGGAUUCUGAUUCCGGCAUUCCGAGGUUCGAAUCCUCGUACCCCAGCCA
UGGGGUAUCGCCAAGCGGUAAGGCACCGGAUUCUGAUUCCGGCAUUCCGAGGUUCGAAUCCUCGUACCCCAGCCA
-
Программа einverted при задании большого threshhold не выдает никакого выравнивания при его понижении до 6 выдает
одно выравнивание первой спирали(отмеченной зеленым). При понижении Gap penality в выравнивании появляються гэпы что немного приближает выравнивание к реальной структуре, но т.к.
выравнивание линейное(т.е. нельзя выравнить участок отсюда потом из другоо места а потом между ними) то эти выравнивания не будут схожи с реальной структурой.
Файлы с выравниванииями:
gap penality - 1
gap penality - 2
gap penality - 3
-
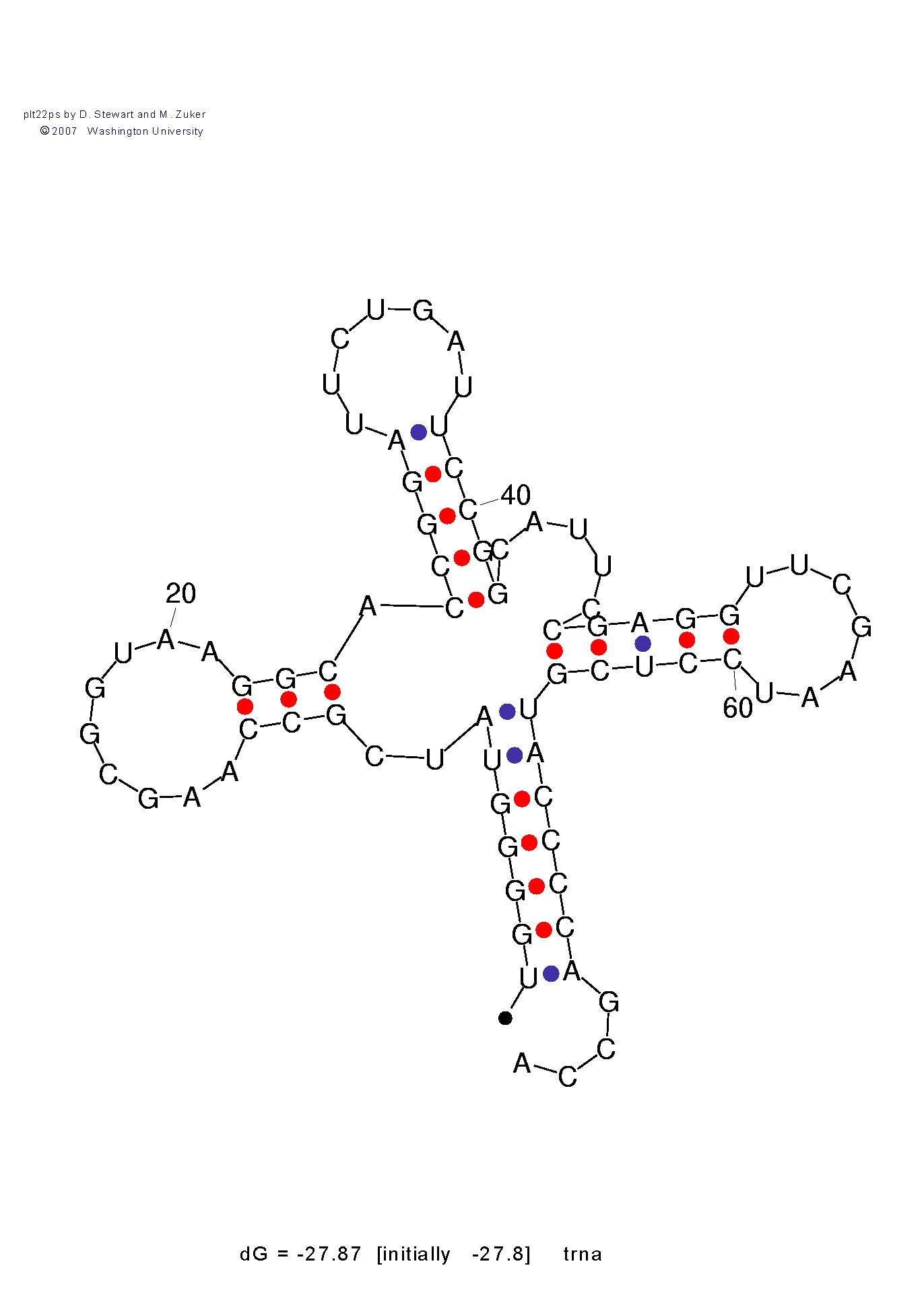
Программа mFold при запуске без параметров выдает структуру не очень похожую на исходную. И только при задании процента P 20. Выдает структуру наиболее близкую к реальной.
Это может происходить от того, что тРНК находиться в комплексе с белком и он ее "изгибает" и ее энергия в этом не самая выгодная с точки зрения отдельной тРНК, но с другой
стороны если учитывать и энегрию белка данная конформация являеться по видимому наиболее выгодной.
Изображение без задания P(справа) и Изображение наиболее близкое к реальной структуре (p=20)(слева):

|
{kind=link}