Выбор домена
Для данной работы выбрала SH2 - Белковый домен SH2, позволяющий содержащим его белкам взаимодействовать с фосфорилированными тирозинами других белков. Белки, содержащие этот домен, обычно участвуют в передаче сигнала рецептору тирозин-киназы. Pfam AC PF00017, Pfam ID SH2. Домен входит в 9081 последовательность из 283 организмов, входит в 379 доменных архитектур, в базе данных PDB находится 390 трехмерных структур. Ниже приведено изображение каталитической субъединицы, содержащий данный домен (рис. 1). Страница домена в Pfam.
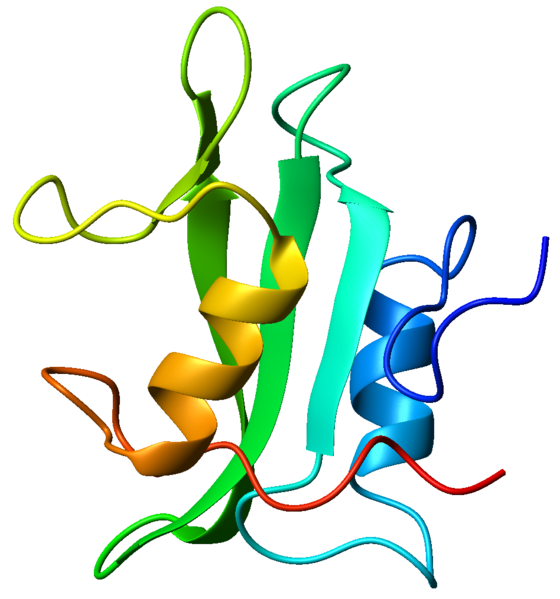
Рисунок 1. SH2, пространственная структура.
С помощью Jalview загрузила выравнивание домена. Ассоциировала структуру 2CR4 c 3BP2_HUMAN. Сделала раскраску по ClustalX и консервативность 13%. Сохранила выравнивание в формате .fasta и .jar.
Выбрала домены для дальнейшей работы.
| домены входящие в архитектуру | схема | количество последовательностей |
| SH3_1, SH2, Pkinase_Tyr |  |
865 |
| PID, SH2 |  |
242 |
Ссылка на список архитектур в Pfam.
Описание доменов:
SH3_1- домен цитоплазматических тирозин-киназ и фосфолипаз, не входящий в их каталитические центры. ссылка в Pfam на домен SH2\3_1 Pkinase_Tyr- домен тирозин-киназны. ссылка в Pfam на домен Pkinase_Tyr PID- домен, взаимодействующий с фосфорилированным тирозином. ссылка в Pfam на домен PID
Выбор таксонов и подтаксонов: Для архитектуры SH2+SH3_1+Pkinase_Tyr было выбрано 12 последовательностей с систематикой Metazoa, Chordata и 13 с систематикой Metazoa, Ecdysozoa,3 с систематикой Metazoa, Platyhelminthes, 4 с систематикой Metazoa Porifera. Для архитектуры PID+SH2 было выбрано 24 последовательностей с систематикой Metazoa, Chordata и 15 с систематикой Metazoa, Ecdysozoa.
Получила таблицу с информацией об архитектуре всех последовательностей, содержащих выбранный домен с помощью скрипта swisspfam_to_xls.py. Составила список последовательностей с указанием доменной архитектуры. Использовала сводную таблицу в Excel: строки – AC последовательностей, столбцы – домены Pfam. Скачала полные записи всех последовательностей из Uniprot, запустила скрипт uniprot_to_taxonomy.py. Перенесла таксономию с основную таблицу. Далее выбрала таксоны, как описано выше.
Выбрала по 25 последовательностей из каждой выбранной выше архитектуры. С помощью скрипта filter_alignment.py оставила только выбранные последовательности из выравнивания. Далее открыла проект в JalView, удалила пустые колонки, создала группы по архитектурам (сверху находятся последовательности, взятые из белков с одним доменом, внизу - последовательности в архитектуре), удалила плохо выровненные участки. Сохранила выравнивание в формате .jar. Ниже привожу изображение выравнивания (ClustalX, консервативноcть 30%) (рис. 2).
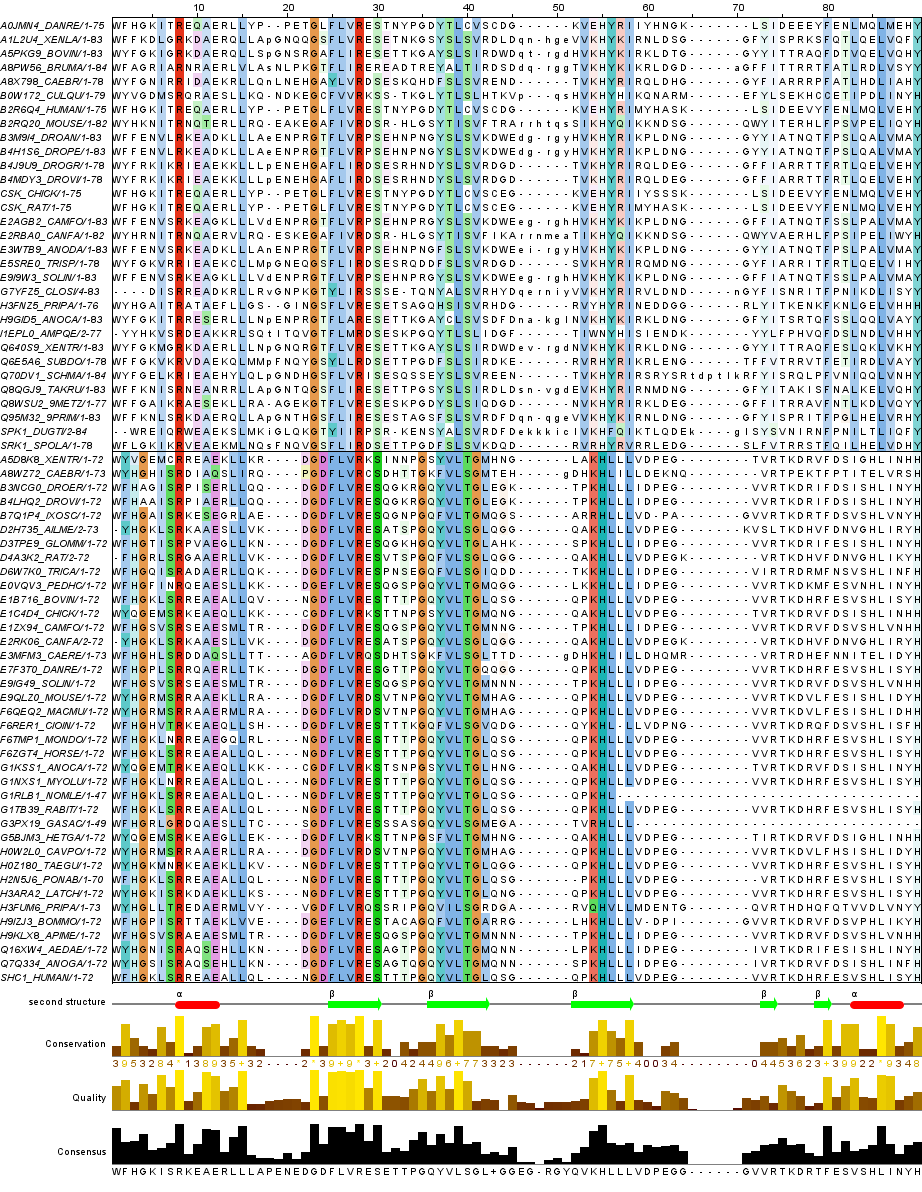
Рисунок 2.Выравнивание отобранных последовательностей домена. Сверху выравнивание двудоменного белка, внизу - трехдоменного.
Исходя из выравнивания, все-таки можно понять, что большая консервативность в значимых участках - альфа-спиралях и бета-тяжах - существует. Наибольшая консервативность наблюдается у трехдоменных белков.