
Сигналы в геноме
Задание 1
Для изучения я выбрал сайт посадки Rho-фактора. Он представляет собой отрезок из 78 нуклеотидов на мРНК с большим отношением C/G (в предложенном алгоритме больше единицы) и равномерно распределенными C (в статье каждые 11-13 нуклеотидов). Rho-фактор, как известно, садится на свой сайт вблизи 3` конца мРНК, перемещается к 3` концу и заставляет РНК-полимеразу диссоциировать от мРНК. Так, как Rho-фактор движется по мРНК медленнее, чем РНК-полимераза, сигнал включает также консенсусную последовательность притормаживания РНК-полимеразы или GC-богатую шпильку чуть дальше к 3` концу (в алгоритме в пределах 150 нуклеотидов), чем сайт связывания Rho.
Адресатом данного сигнала является Rho-фактор, от которого ожидается связывание с представленным сайтом. Предположу, что сила данного сигнала высока, так как терминация транскрипции крайне важный процесс, поэтому, было бы логично, если бы Rho-фактор, связывался со своим сайтом, как только предоставлялась бы такая возможность.
Как только я посмотрел на код уважаемого доктора Ди Сальво, мне сразу стало понятно, что этот человек незнаком (или не был знаком на момент написания статьи) с современными производственными стандартами написания кода. Кроме того, мне кажется, господин Ди Сальво слишком вольготно распоряжается числами. Я могу понять откуда взялась длина сайта связывания - 78 нуклеотидов, но почему C богатый регион ищется в пределах 150 нуклеотидов от старта сайта непонятно. Мне кажется, его исследование достаточно невремязатратное, и могло бы быть выполнено куда лучше. На его месте я бы подобрал эти гиперпараметры опираясь на секвенирование транскриптомов с использованием ингибитора Rho.
Я бы безусловно не поленился написать этот алгоритм сам, будь этот семестр посвободнее: я реализовал его частично, и тем не менее, по моему, вполне достаточно для наших задач. Я реализовал поиск равномерно расположенных C в последовательностях обогащенных C и обедненных G, с помощью системы непересекающихся множеств выделил подходящие сайты, из них выбрал последний встречающийся. Я реализовал поиск только консенсусной последовательности притормаживания в конце мРНК. Я позволил себе несколько отойти от алгоритма из статьи, в работе Ди Сальво указано, что консенсусная последовательность притормаживания РНК-полимеразы находится ровно на конце мРНК, но в его алгоритме, она ищется не на конце мРНК а в окне размером 150 нуклеотидов от 3` конца сайта посадки. Я же искал эту последовательность, ровно как указано в статье: на конце мРНК. Среди полного аннотированного генома E.Coli я нашел 11 мРНК с таким типом терминации. Привожу названия генов и сайты ниже:
pcnB CAATCCCGAAACGTCTGACGACATTAACCCGCGATATCTGGCAGTTGCAGTTGCGTATGTCCCGTCGTCAGGGTAAACG
metN CGCATCCGAAAACGCCGCTGGCGCAGAAGTTTATTCAGTCGACCCTGCATCTGGATATCCCGGAAGATTACCAGGAACG
ybeZ TCACGCCATCGAAGTGCTGGCCGATGTCGAAGAGATCAGCTTTAACTTCTTCCACAGCGAAGACGTGGTTCGTCACCCT
narZ CCAACGGCGCGCTGACTGCCCGCGCGGTGGTCAGCCAACGTGTACCGCCGGGCATGACCATGATGTATCACGCCCAGGA
astD CCAGTACCGCGCCATTCGGCGGCATTGGTGCTTCCGGTAACCATCGCCCCAGCGCCTGGTATGCCGCAGATTACTGCGC
wcaB GCGGCCCCGCTGCTGGTGCTGTATCGCATTATCACCGAATGCTTTTTCGGTTATGAAATCCAGGCTGCCGCGACCATTG
eutH CGACCACTTAGGCTTCGCCGCTGCCAACATGAACGCCATGATCTTCCCGATGATTGTCGGCAAGTTGATCGGCGGCGTA
csdA CGCTGGCGAAACGTCCCGGCTTTCGTTCATTCCGCTGCCAGGATTCCAGCCTGCTGGCCTTTGATTTTGCTGGCGTTCA
yhfA CGCCTGTTTACGCACATTAATCTGCATTTTATCGTCACCGGTCGCGACCTGAAAGACGCAGCGGTTGCGCGTGCGGTTG
fabR CGGTAATAATCCTAACGCCTTCCGGTTATTATTGCGGGAACGCTCCGGCACCTCCGCTGCGTTTCGTGCCGCCGTTGCG
frdA AAGAGTCCCGCGGCGCGCACCAGCGTCTGGACGAAGGTTGCACCGAGCGTGACGACGTCAACTTCCTCAAACACACCCT
Как мы видим, программа отработала штатно, в каждой последовательности мы видим равномерно распределенные C, и некоторую обедненность G в сочетании с обогащенностью C.
Источник: Di Salvo, M., Puccio, S., Peano, C. et al. RhoTermPredict: an algorithm for predicting Rho-dependent transcription terminators based on Escherichia coli, Bacillus subtilis and Salmonella enterica databases. BMC Bioinformatics 20, 117 (2019). https://doi.org/10.1186/s12859-019-2704-x
Задание 2
Для изучения я выбрал последовательность Шайна-Дальгарно E. coli, которые выделил из выравнивания десяти нуклеотидов до ATG, в качестве теста выделил участки с такими же координатами из B. subtilis. Получилась следующая PWM:
0 1 2 3 4 5 6
A: 1.73 -2.14 -1.94 1.34 0.53 0.67 0.47
C: -4.92 -4.60 -5.90 -1.85 -2.01 -1.08 -0.22
G: -0.66 1.87 1.86 -0.86 0.72 -0.25 -0.50
T: -4.08 -4.88 -4.43 -0.57 -0.59 0.16 0.09
IC выравнивания равно 5.155. Гистограмма весов для положительных, отрицательных и тестовых примеров приведена ниже:
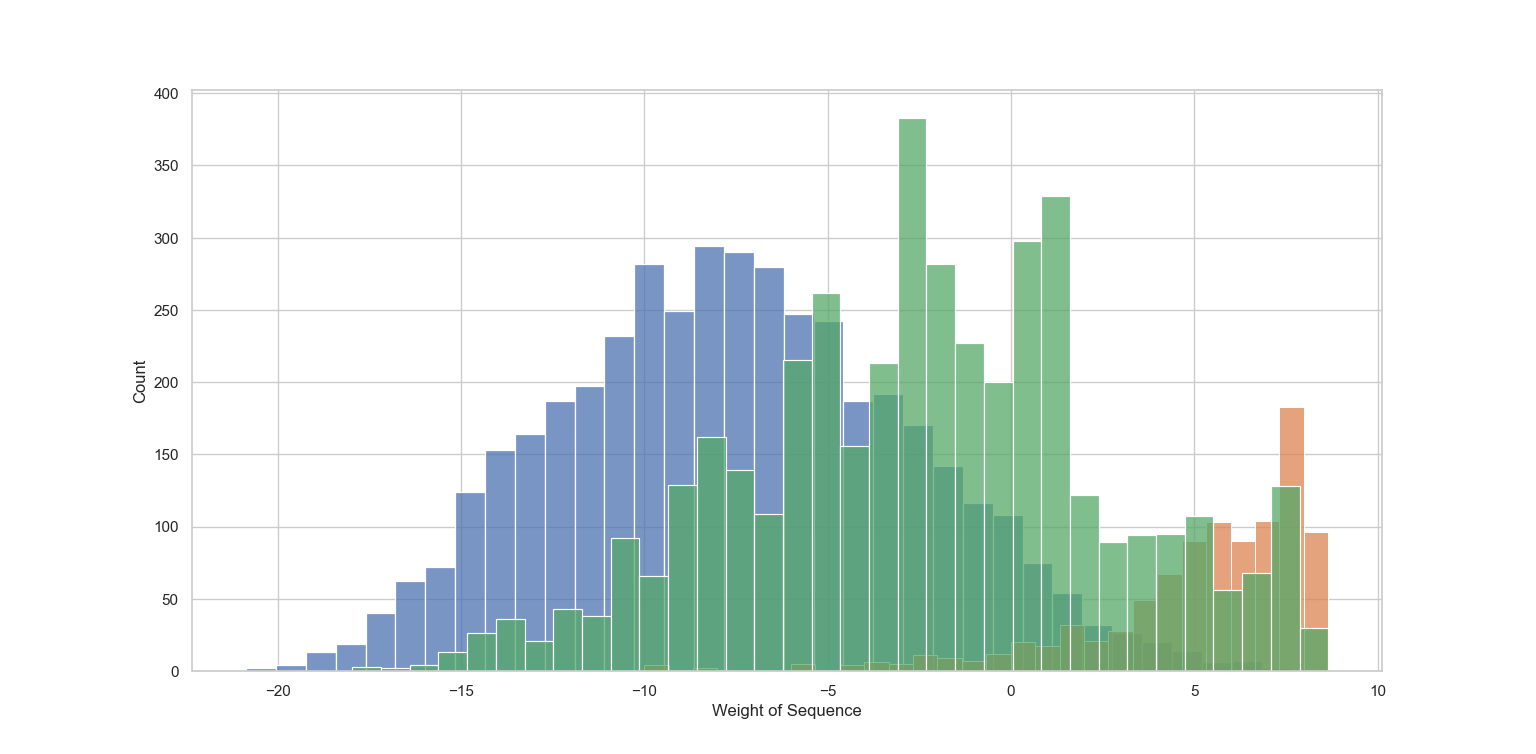
В качестве порога веса для положительного предсказания я выбрал 3. Получились результаты представленные ниже:
| Удовлетворяют PWM | Не удовлетворяют PWM | |
|---|---|---|
| Положительные примеры | 798 | 170 |
| Отрицательные примеры | 67 | 4238 |
| Тестовые примеры | 589 | 3648 |
Видимо, последовательности Шайна-Дальгарно несколько варьируются у двух этих видов либо по составу, либо по положению относительно старт-кодона.