Комплексы ДНК-белок. Вторичная структура тРНК.
Предсказание вторичной структуры РНК осуществлялось двумя методами: с помощью поиска инвертированных повторов (программа einverted из EMBOSS) и с помощью алгоритма Зукера, который реализуется с помощью программы RNAfold из пакета Viena RNA Package.
Для начала pdb файл со структурой моей РНК (PDB ID 1qu2) нужно было конвертировать в старый формат с помощью программы remediator, чтобы потом можно было с помощью find_pair получить необходимые данные о вторичной структуре. Но это было сделано уже в прошлом практическом занятии, поэтому возьму уже готовый файл 1qu2_old.pdb. Описание в этом файле и будет считаться за "реальную структуру" тРНК.
Нуклеотидная последовательность РНК для einverted была получена так же с RCSB.org в виде fasta-файла. Для работы einverted были произведены попытки подобрать некоторые параметры для получения предсказания, наиболее близкого к реальной структуре, но спустя 3 попытки так и не удалось, к сожалению, этого получить. Дальше приведу параметры, которые были выставлены в эти 3 попытки:
-gap 0 -threshold 0 -match 20 -mismatch -4
-gap 20 -thr 20 -match 6 -mis -8
-gap 12 -thr 0 -match 3 -mis -12
По итогу мы получили файл invertedseq.inv, в котором находится информация о найденных комлиментарных парах.
На рисунке 1 изображен результат работы алгоритма Зукера c первыми параметрами, приведенными выше.
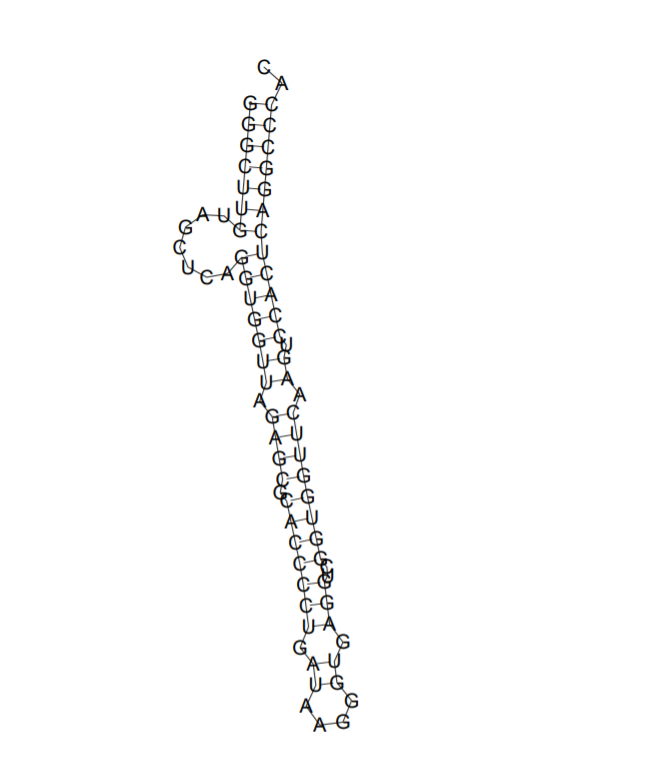
В таблице 1 приведены результаты сравнения реальной вторичной структуры тРНК и двух предсказанных (с помощью алгоритма Зукера и с помощью einverted).
| Участок структуры | Позиции в структуре (find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания
по алгоритму Зукера (RNAfold) |
|---|---|---|---|
| Акцепторный стебель | 5'_1-7_3'; 5'_66-72_3`; 7 пар | 4/7 и 3 лишние | 7/7 |
| D-стебель | 5'_10-12_3'; 5'_23-25_3`; 3 пары | 0/3 | 3/3 |
| T-стебель | 5'_49-52_3'; 5'_62-65_3'; 4 пары | 0/4 | 4/4 |
| Антикодоновый стебель | 5'_27-31_3'; 5'_39-43_3'; 5 пар | 0/5 | 5/5 |
| Общее число канонических пар нуклеотидов | 19 | 4 | 19 |
По итогу мы видим, что алгоритм Зукера может полностью предсказать структуру тРНК.
В данном задании исследовались контакты между ДНК и белка в PDB-модели 1efa.
Скачать скрипт можно по ссылке.
С помощью следующего скрипта был произведен поиск белковых контактов.
В таблице 2 приведено число различных контакотов белка с разными "частями" нуклеиновой кислоты.
| Контакты атомов белка с | Полярные | Неполярные | Всего |
|---|---|---|---|
| остатками 2'-дезоксирибозы | 1 | 42 | 43 |
| остатками фосфорной кислоты | 21 | 20 | 41 |
| остатками азотистых оснований со стороны большой бороздки | 5 | 26 | 31 |
| остатками азотистых оснований со стороны малой бороздки | 24 | 41 | 65 |
По результатам, приведенным в таблице 2, можно заметить, что количество неполярных ДНК-белковых контактов существенно превосходит количество полярных в остатках 2'-дезоксирибозы и в атотистых основаниях как со стороны малой, так и со стороны большой бороздок.
Контакты также позволяет описать программа nucplot. Результаты работы данной программы представлены на рис.2.
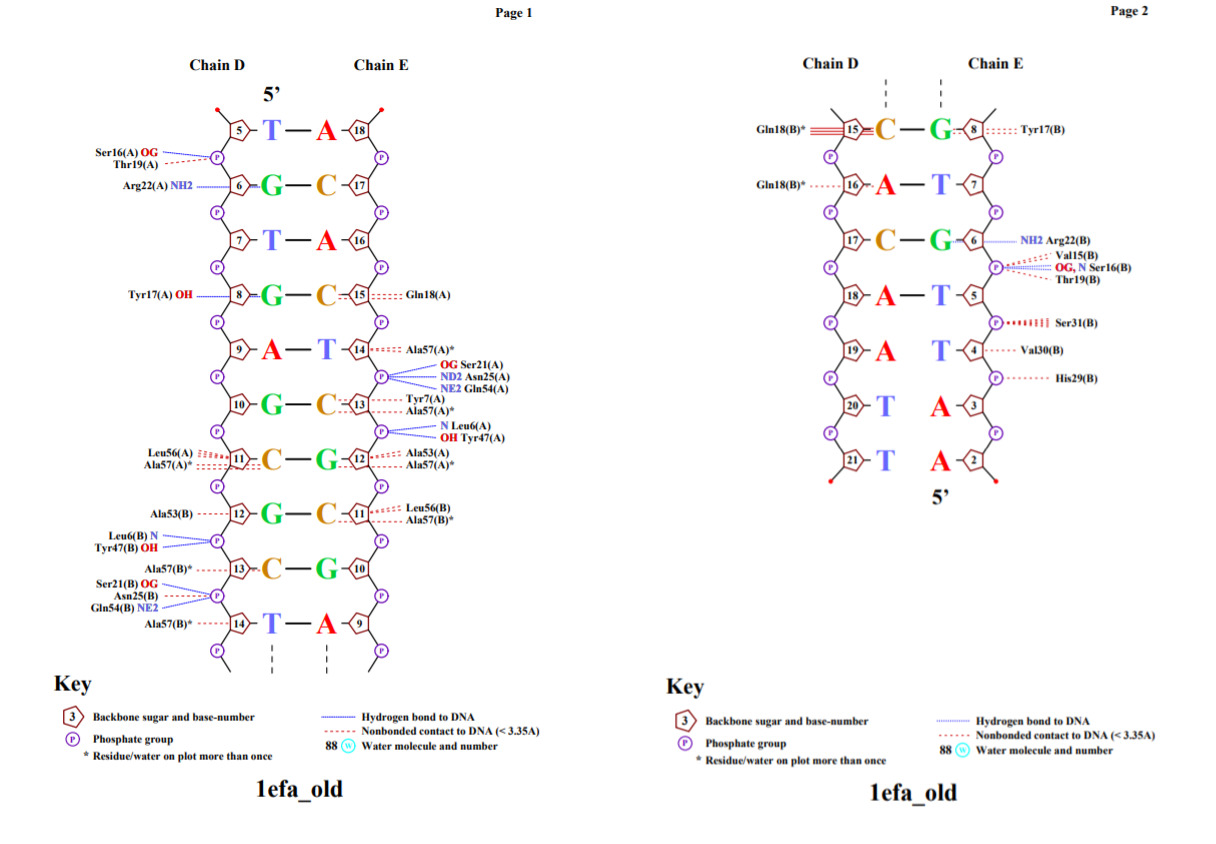
Для дальнейшей работы был выбран аминокислотный остаток Ala57(A) как имеющий наибольшое число контактов (4) с ДНК на схеме, приведенной на рис. 2.
Ala57(A) вступает во взаимодействия с атомами азотистых оснований C(11) D-цепи, С(13) и G(12) Е-цепи, а также с атомами дезоксирибозы при Т(14) Е-цепи.
Также, по моему мнению, этот аминокислотный остаток является наиболее важным для распознавания последовательности ДНК, т.к. он не тольок имеет больше всего контактов с структурой, но и эти контакты принадлежат нескольким азотистым основаниям. Рис. 3 иллюстрирует приведенный пример.
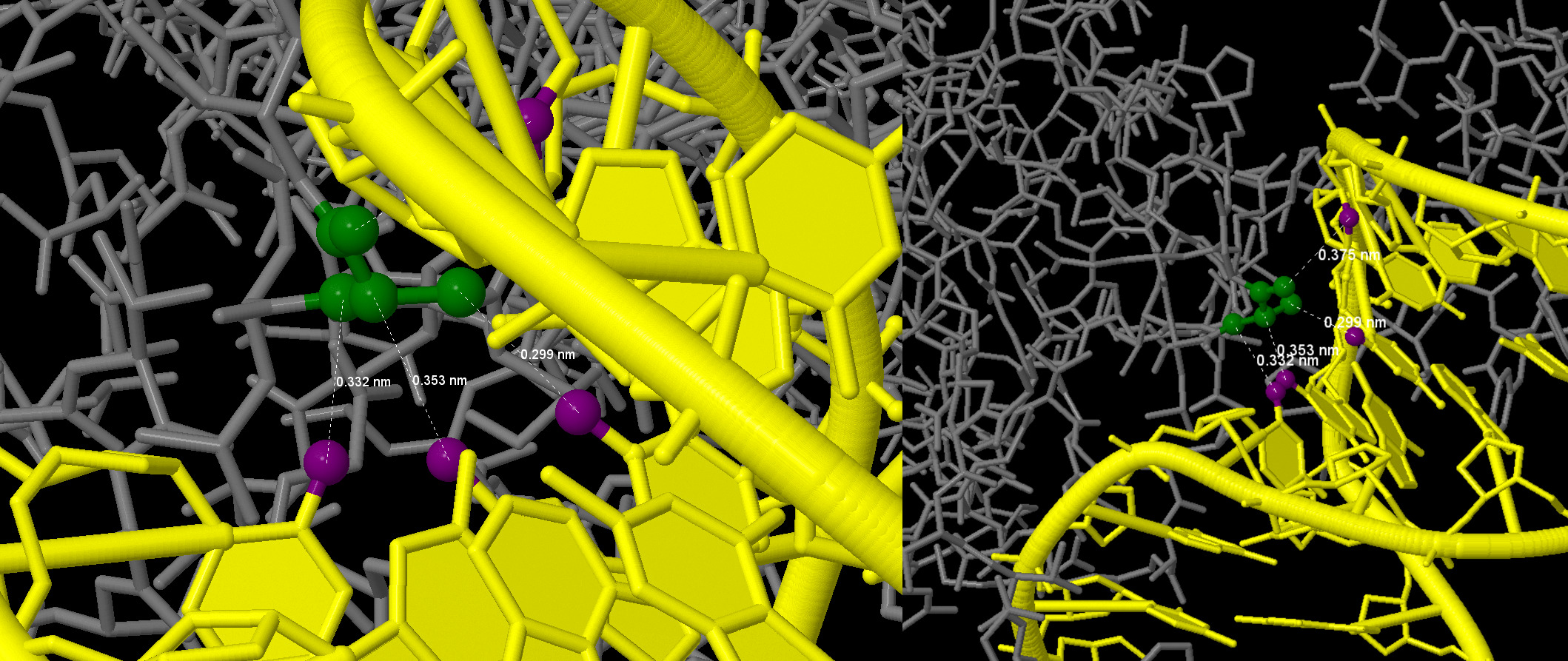
Желтым цветом показана ДНК, зеленым - атомы Ala57(A),
фиолетовым - атомы азотистых оснований и дезоксирибозы,
вступившие в связь с аминокислотным остатком.