Часть 1. Подготовка чтений
Для того, чтобы выполнить это задание, были выданы два файла:
°
файл с одноконцевыми чтениями экзома 5 хромосомы человека chr5.fastq;
°
файл с последовательностью этой хромосомы из генома со сборкой версии hg19 chr5.fasta
1. Анализ качества чтений с помощью программы FastQC для исходного,
chr5.fastq, файла .
Использованная команда - "fastqc chr5.fastq".
Файл на выходе -
chr5_fastqc.html, который содержал
графические данные и архив chr5_fastqc.zip(изображения+текстовые файлы).
2. Очистка чтений с помощью программы Trimmomatic.
Требовалось отрезать с конца каждого чтения нуклеотиды с качеством ниже 20,
но оставить только чтения длиной не меньше 50 нуклеотидов.
Разбор примененных функций:
° TRAILING - удаляет с конца чтения нуклеотиды с качеством ниже указанного.
Например,"TRAILING:20" = удалению нуклеотидов с качеством ниже 20.
° MINLEN - удаляет чтения с длиной меньше указанного числа.
Например, "MINLEN:50" = подразумевает, что останутся только чтения, длина
которых не меньше 50.
Использованная команда - "java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr5.fastq chr5_out.fastq TRAILING:20 MINLEN:50".
Файл на выходе -
chr5_out.fastq
3. Анализ качества чтений с помощью программы FastQC для полученного файла
chr5_out.fastq.
Использованная команда - "fastqc chr5_out.fastq".
Файл на выходе -
chr5_out_fastqc.html.
4. Сравнить выдачу
chr5_out_fastqc.html с выдачей
chr5_out.fastq.
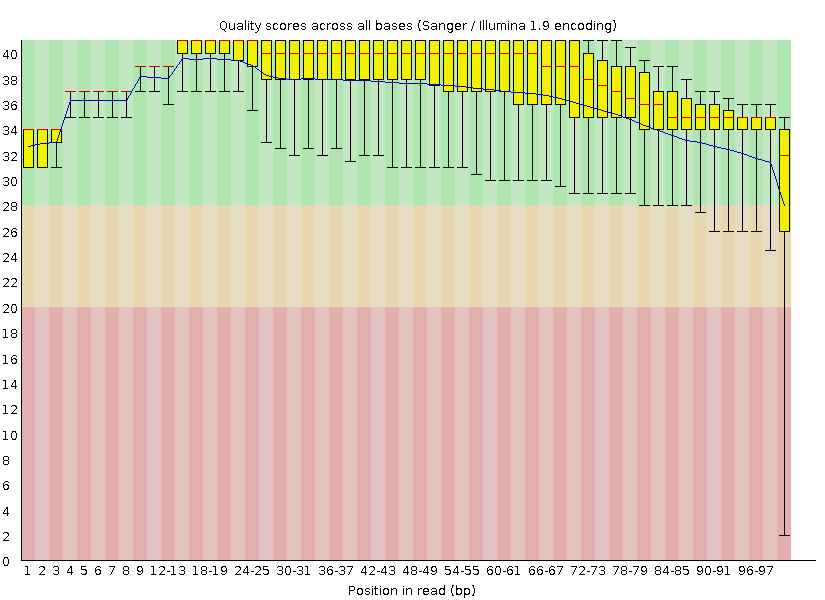
До очистки:
° число чтений = 8208.
° длина чтений = 41-100.
Per base quality до чистки:
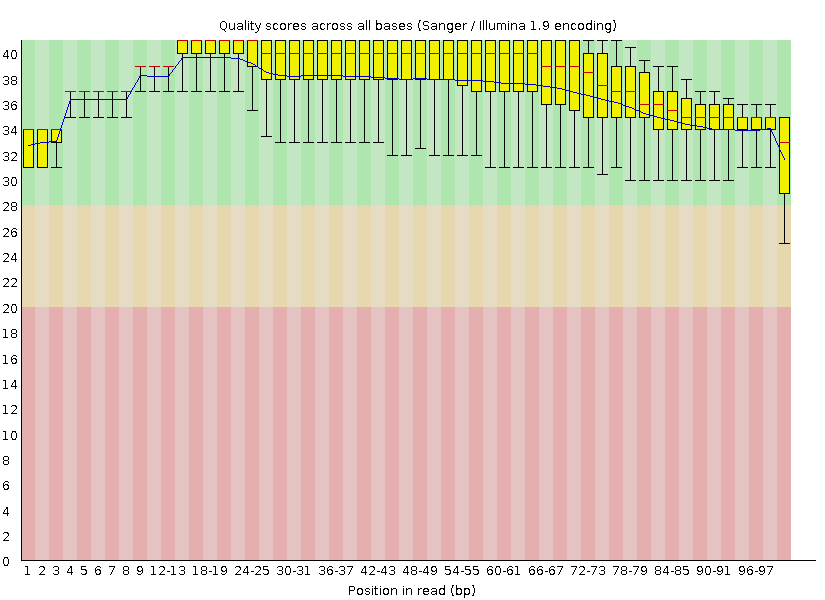
После очистки:
° число чтений = 8114.
° длина чтений = 50-100.
Per base quality после чистки:
Часть 2. Картирование чтений
5. Откартирование чтений с помощью программы BWA происходит в 2 этапа:
° Индексирование референсной последовательности:
Использованная команда - "bwa index chr5.fasta"
Время работы программы: 368.304 sec.
° Построение выравнивания прочтений и референса в формате .sam:
Использованная команда - "bwa mem chr5.fasta chr5_out.fastq > chr5.sam".
Файл на выходе -
chr5.sam.
Время работы программы: 0.928 sec.
6. Анализ выравнивания происходит в 4 этапа:
° Перевод полученного файла в бинарный формат:
Использованная команда - "samtools view chr5.sam -bo chr5.bam".
Файл на выходе -
chr5.bam.
° Отсортировка выравниваний чтений с референсом(полученный ранее bam-файл)
Использованная команда - "samtools sort chr5.bam -T pr13.txt -o chr5_sort.bam".
Файл на выходе -
chr5_sort.bam.
° Индексирование отсортированного bam-файла.
Использованная команда - "samtools index chr5_sort.bam".
Файл на выходе -
chr5_sort.bam.bai.
° Анализ количества чтений, откартированных на геном:
Использованная команда - "samtools idxstats chr5_sort.bam > pr13_out.txt"
Файл на выходе -
pr13_out.txt - в нем указаны название
последовательности, длина последовательности (180915260) и число картированных чтений (8113). Таким образом, только одно чтение не было картировано.
Часть 3. Картирование чтений
7. Поиск SNP и инделей проходит в 3 этапа:
° Создание файла с полиморфизами
Использованная команда -"samtools mpileup -uf chr5.fasta chr5_sort.bam -o chr5.bcf".
Файл на выходе -
chr5.bcf.
° Создание файла со списком отличий между референсом и чтениями
Использованная команда - "bcftools call -cv chr5.bcf -o chr5.vcf".
Файл на выходе -
chr5.vcf
° Нахождение и описание трех полиморфизмов из полученного файла
chr5.vcf
Немного о самом файле:
→ в его составе 32 полиморфизма;
→ 2 вставки;
→ 2 делеции;
→ 28 SNP.
8. Аннотация SNP
° Для начала необходимо конвертировать и аннотировать полученный файл без инделей
chr5_without_indel.vcf
с помощью скриптов convert2annovar.pl и annotate_variation.pl.
Использованная команда - "perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 chr5_without_indel.vcf -outfile
chr5.annovar"
Файл на выходе -
chr5.annovar
Далее с файлом
chr5.annovar можно произвести аннотации SNP с помощью скрипта
annotate_variation.pl.
→ Refgene - gene-based annotation
Использованная команда - "perl /nfs/srv/databases/annovar/annotate_variation.pl -out chr5.refgene -build hg19 chr5.annovar /nfs/srv/databases/annovar/humandb/"
Файлы на выходе:
°
chr5.refgene.log
°
chr5.refgene.variant_function
°
chr5.refgene.exonic_variant_function
16 полиморфизмов оказались гомозиготными, а 12 - гетерозиготными. Также в выходном файле указаны названия генов, в которые попали полиморфизмы:
° IL7R
° CAPSL
° HMGCR
→ dbSNP (Single Nucleotide Polymorphism Database)
Использованная команда - "perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr5.dbsnp -build hg19 -dbtype snp138 chr5.annovar /nfs/srv/databases/annovar/humandb/"
°
chr5.dbsnp.hg19_snp138_dropped
°
chr5.dbsnp.hg19_snp138_filtered
°
chr5.dbsnp.log
5 хромосом 24 SNP имеют rs, а 4 - не имеют. Все полиморфизмы, которые расположены на экзонах, имеют аннотацию в базе данных dbSNP.
→ GWAS (Genome-Wide Association Studies)
Использованная команда - "perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -build hg19 -out chr5.gwas -dbtype gwasCatalog chr5.annovar /nfs/srv/databases/annovar/humandb/"
°
chr5.gwas.hg19_gwasCatalog
°
chr5.gwas.log
→ 1000 Genomes
Использованная команда - "perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out chr5.1000g -dbtype 1000g2014oct_all -buildver hg19 chr5.annovar /nfs/srv/databases/annovar/humandb/"
°
chr5.1000g.hg19_ALL.sites.2014_10_dropped
°
chr5.1000g.hg19_ALL.sites.2014_10_filtered
°
chr5.1000g.log
→ ClinVar
Использованная команда - "perl /nfs/srv/databases/annovar/annotate_variation.pl chr5.annovar -filter -dbtype clinvar_20150629 -buildver hg19 -out chr5.clinvar /nfs/srv/databases/annovar/humandb/"
°
chr5.clinvar.hg19_clinvar_20150629_dropped
°
chr5.clinvar.hg19_clinvar_20150629_filtered
°
chr5.clinvar.log