Задание 1. Прочитать последовательность ДНК на основании данных, полученных из капиллярного секвенатора по Сэнгеру. Составить отчёт о проблемах при чтении хроматограмм.
Исходные файлы:
° Прямая цепь
° Обратная цепь
Для просмотра хроматограмм и редактирования автоматического прочтения использовалась программа Chromas (Lite).
В программе были открыты оба исходных файла, при этом файл с обратной цепью был переведен в комплементарную цепь с помощью команды Edit ->
Reverse+Complement. Затем для обоих последовательностей был настроен одинаковый масштаб по горизонтали и было произведено выравнивание
двух последовательностей с использованием поиска подслов Find.
Таблица 1. Координаты нечитаемых 5'- и 3'-концов (по прямой цепи).

Оценка хроматограмм:
° Прямая цепь:
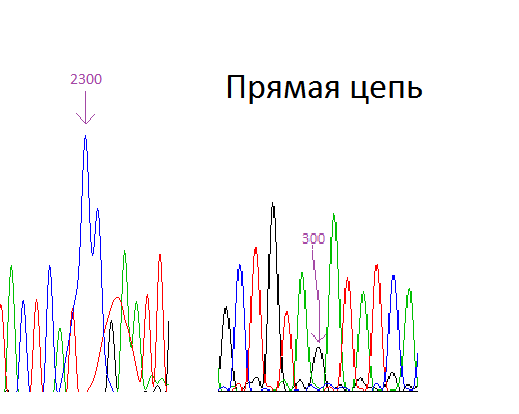
Средняя сила сигнала ≈ 1000-1200. Шум распределен равномерно.
Сигнал преобладает над шумом ≈ в 15 раз. Разброс значений сигнала наблюдается ≈ от 300 до 2300, следовательно,
это наименьшее и наибольшее значения на имеющейся хроматограмме. Сила сигнала не зависит от имеющегося нуклеотида
(пример: максимальная сила сигнала принадлежит цитозину, но в тоже время для данного нуклеотида имеются пики с невысоким
значением; подобные примеры наблюдаются неоднократно).
° Обратная цепь:
Сигнал преобладает над шумом в такой же пропорции, как и в прямой цепи.
Средняя сила сигнала ≈ 1100-1200. Шум также распределен равномерно.
Разброс значений сигнала наблюдается ≈ от 340 до 2450, следовательно, это наименьшее и наибольшее значения на имеющейся хроматограмме.
Сила сигнала не зависит от имеющегося нуклеотида по той же причине, что имелась и у прямой цепи.


Обоснование решений для проблемных нуклеотидов:
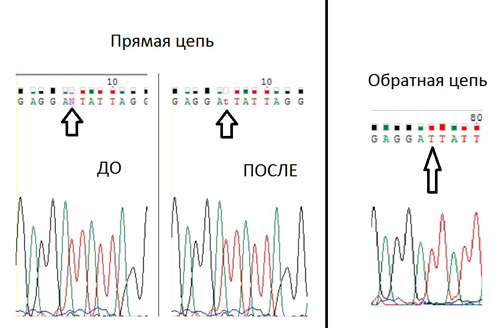
На рисунках показаны некоторые из произведенных мною замен.
#1
° Выделенный программой нуклеотид был ею не определен в виду наложения сигналов друг на друга. Ориентировашись по обратной цепи, заменен на необходимый.



Задание 2. Пример плохой хроматограммы
