Ресеквенирование и поиск полиморфизмов у человека
Используем пайплайн, чтобы найти и охарактеризовать полиморфизмы в ридах пациента. Я работал с хромосомой 19.
Процесс работы
Основные этапы работы оформлены в виде сценариев для bash. Вот порядок команд, соответствующий изложению заданий (опущен этап копирования файлов сценариев):
- cd /nfs/srv/databases/ngs/
- mkdir russal2010
- cp Human/chr19.fasta russal2010/
- cd russal2010
- mkdir index
- mkdir reads
- hisat2-build chr19.fasta index/chr19.idx
- cp -r ../Human/reads/chr19.fastq reads/
- ./fastqc.sh
- ./trim.sh
- ./trim_fastqc.sh
- ./mapping.sh
- ./analysis.sh
- ./snp.sh
- ./variations.sh > variations.o 2> variations.e
Задействованные файлы:
Смысл основных команд описан в таблице 1.
| Команда | Смысл |
|---|---|
| hisat2-build chr19.fasta index/chr19.idx | Создаём индекс HISAT2 по файлу chr19.fasta в папке ./index/ с базовым именем chr19.idx. |
| fastqc $reads | Запускаем FastQC. |
| $trim SE -phred33 $reads ${trim_reads} TRAILING:20 MINLEN:50 | Триммирование: убираем с конца нуклеотиды с качеством прочтения ниже 20, а затем отбрасываем слишком короткие (в том числе укоротившиеся после предыдущей операции) чтения. |
| hisat2 --no-spliced-alignment --no-softclip -x $idx -U ${trim_reads} -S ${wdir}/map/chr19_aln.sam 2> ${wdir}/map/out.txt | Осуществляем картирование, не допуская сплайсинга и софтклиппинга (обрезки концов не полностью лёгших ридов). Опцией -x предоставляем путь к индексу, также вывод (stderr) сохраняем в map/out.txt. |
| samtools view -b $samfile -o $bamfile | Переводим файл .sam в бинарный формат. |
| samtools sort -o $sortedbam -O bam -T ./'sorttemp' $bamfile | Сортируем файл .bam, предоставляя samtools шаблон пути для временных файлов (-T). |
| samtools index $sortedbam | Индексируем сортированный файл .bam. |
| samtools flagstat $sortedbam > ${flag_out} | Собираем базовую статистику по картированию, альтернативную выводу HISAT2. |
| samtools faidx $ref | Создаём короткую характеристику референнса в табличном виде (включает название, идентификатор, длину последовательности, номер первого нуклеотида — какой по счёту байт в файле, число нуклеотидов и байтов на строку). |
| samtools mpileup -u -f $ref $sortedbam -o $ref\.pile > snp_out.txt 2> snp_err.txt | Создаём .bcf файл с полиморфизмами, сохраняя вывод (stdout и stderr) в файлы. -f указывает, что референс был в формате fasta, -u — что вывод в .bcf, а не .vcf. |
| bcftools call -c -v -o $ref\.pile_indels.vcf $ref\.pile | Переводим .bcf в .vcf с инделями. -c — берём только «консенсусные» SNP (редкие не будут учтены), -v — в вывод попадут только сами сайты SNP. |
| bcftools call -c -v -V indels -o $ref\.pile.vcf $ref\.pile | То же, без инделей (опция -V indels). |
| convert2annovar.pl -format vcf4 ${ref}.pile.vcf > ${ref}.avinput | Готовим файл .vcf к применению annovar. |
| annotate_variation.pl -out $refg -build hg19 -dbtype refGene ${ref}.avinput ${avdir}/humandb.old/ | Эта и подобные команды — аннотация SNP по указанной базе. -out — опция для указания пути к файлу вывода с аннотацией,-build — опция для уточнения версии сборки генома человека, -dbtype — по какой базе аннотируем; далее указан путь к файлу с неаннотированными SNP и к папке с базой для аннотации. |
По исходному отчёту FastQC смотрим число чтений в исходном файле (5524). На рис. 1 приведена оценка их качества.
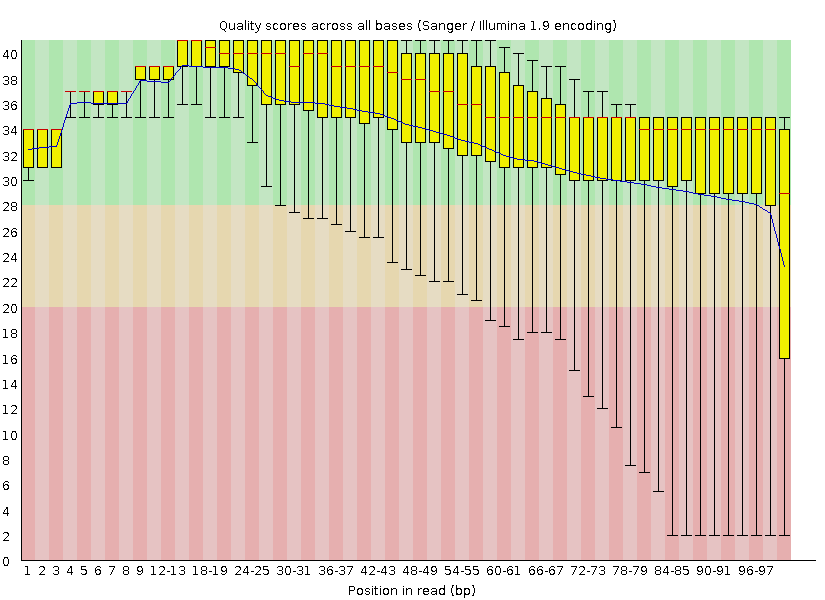
После тримминга чтений осталось 5227 (отчёт). Длина в среднем сократилась незначительно (см. Sequence Length Distribution), а вот качество выросло (приведено на рис. 2). И отсечено, получается, было более 5 % чтений. Эта доля не выглядит пренебрежимо малой, поэтому триммирование представляется оправданным.
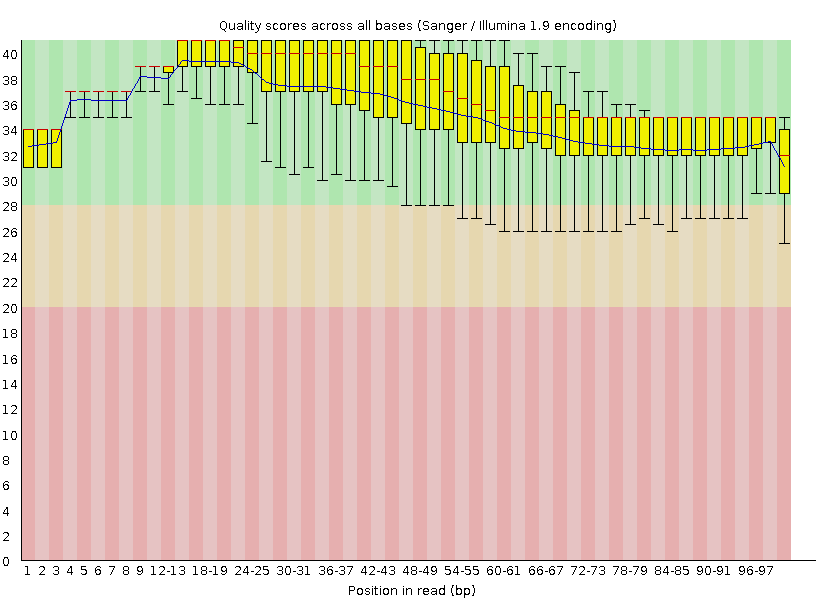
Вывод программы картирования сообщает, что картировано 99,60 % чтений. Это практически все — напрашивается вывод, что чтения ложатся хорошо. Теперь рассмотрим подробнее файл .vcf с описанием полиморфизмов. Опишем три экземпляра из него (см. табл. 2).
| Номер примера | Координата | Тип | Референс | Чтения | Покрытие | Качество |
|---|---|---|---|---|---|---|
| 1 | 17247439 | замена | G | C | 42,76 | 2 |
| 2 | 17313178 | вставка | a | aT | 78,47 | 13 |
| 3 | 18304700 | замена | A | G | 222 | 36 |
Всего было 87 SNP и 6 инделей. Данные о распределении качества прочтений полиморфизмов и их покрытии получены с помощью Excel и приведены на рис. 3–4 (гистограммы).
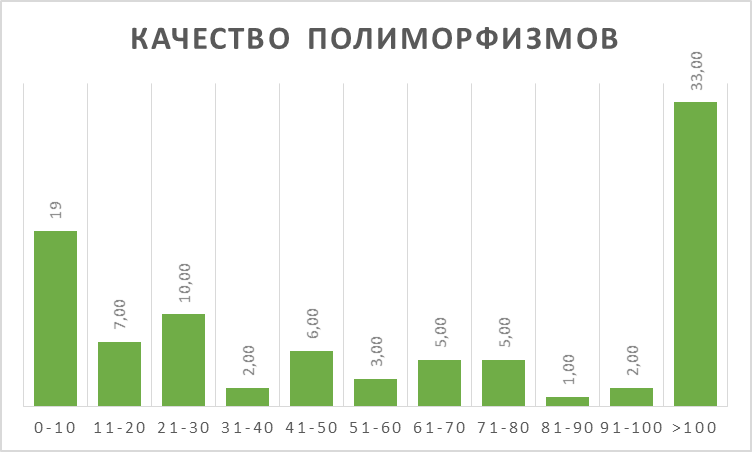
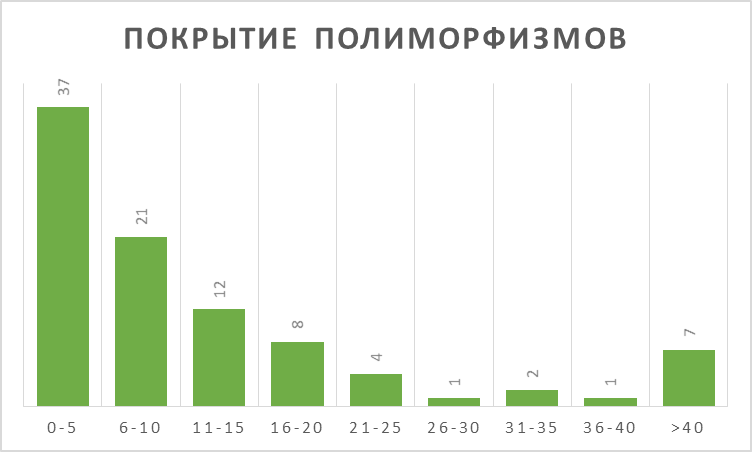
Более четверти полиморфизмов прочитаны с сомнительным качеством (ниже 20) и более трети имеют покрытие на уровне считанных единиц. Это говорит о в целом невысоком качестве выборки, хотя есть и хорошие высокопокрытые образцы (как второй и третий в таблице).
Для аннотации SNP без инделей была применена опция -V indels bcftools. (Файл .vcf с инделями больше не использовался). База данных refseq в annovar делит мои SNP на 3 группы: exonic (12 шт.), intronic (71 шт.), UTR3 (4 шт., соответствуют 3'–нетранслируемому участку). Это отражено в файле .variant_function. Гены, в которые попали полиморфизмы: MYO9B, MPV17L2, TOMM40 (указаны там же). "К каким нуклеотидным заменам привели SNP" — вопрос риторический, потому что сами SNP заменами и являются; тем не менее, ответ существует. Нуклеотидные замены в генах для UTR3 перечислены во всё том же файле, а в экзонах (вместе с аминокислотными заменами) — в файле .exonic_variant_function. Перечислим их все списком (префиксом c снабжаются нуклеотидные замены, префиксом p — аминокислотные; звёздочкой помечаются замены в нетранслируемом регионе):
NM_004145:c.*555A>T,NM_001130065:c.*972A>T
NM_032683:c.*98G>A
NM_032683:c.*243C>T
NM_006114:c.*262G>A,NM_001128917:c.*262G>A,NM_001128916:c.*262G>A
NM_001130065:exon10:c.G1632T:p.L544L,MYO9B:NM_004145:exon10:c.G1632T:p.L544L
NM_001130065:exon13:c.C2025T:p.I675I,MYO9B:NM_004145:exon13:c.C2025T:p.I675I
NM_001130065:exon21:c.T3031G:p.S1011A,MYO9B:NM_004145:exon21:c.T3031G:p.S1011A
NM_001130065:exon22:c.T3204C:p.G1068G,MYO9B:NM_004145:exon22:c.T3204C:p.G1068G
NM_001130065:exon22:c.T3795C:p.P1265P,MYO9B:NM_004145:exon22:c.T3795C:p.P1265P
NM_001130065:exon28:c.C4716T:p.V1572V,MYO9B:NM_004145:exon28:c.C4716T:p.V1572V
NM_001130065:exon32:c.T5078C:p.V1693A,MYO9B:NM_004145:exon32:c.T5078C:p.V1693A
NM_001130065:exon35:c.A5526G:p.S1842S,MYO9B:NM_004145:exon35:c.A5526G:p.S1842S
NM_032683:exon2:c.A214G:p.M72V
NM_032683:exon4:c.C492T:p.P164P
NM_001128917:exon2:c.T339C:p.F113F,TOMM40:NM_001128916:exon3:c.T339C:p.F113F,TOMM40:NM_006114:exon3:c.T339C:p.F113F
NM_001128917:exon3:c.C393T:p.F131F,TOMM40:NM_001128916:exon4:c.C393T:p.F131F,TOMM40:NM_006114:exon4:c.C393T:p.F131F
Чтобы узнать, сколько SNP имеют rs, надо посмотреть на результат работы с базой snp138. В этом файле 77 строк, значит, столько SNP и имело rs. О частоте скажет база 1000genomes (второй столбец файла: числа лежат в диапазоне от 0,0002 до 0,7808; среднее 0,4213, медиана 0,5198). Клиническая аннотация — GWAS. (Аннотация.) По ней, имеется полиморфизм, ассоциированный с ростом, так же, как и полиморфизмы, связанные с рассеянным склерозом, метаболическим синдромом, сердечно-сосудистыми заболеваниями, долголетием, уровнем холестерина и болезнью Альцгеймера (вот про долголетие описано очень широко).
Практикум был очень длинным и запутанным, особенно в части с аннотацией по базам данных. Они лежали в разных папках, а одна из них (1000g2014oct_all) и вовсе должна была быть вызвана не совсем так, как оговорено (сказано про версию '1000g2014oct'). Тем не менее результаты есть, и это не может не радовать.