Анализ данных РНК-секвенирования
Здесь чтения РНК картируются на референс и подсчитывается, куда они попали.
Процесс работы
Основные этапы работы оформлены в виде сценариев для bash. Для приведём команды из них и отдельно список инициализированных переменных. Файл с переменными можно скачать, команды и объяснения приведены в таблице 1.
| Команда | Смысл |
|---|---|
| fastqc ${before} | Запускаем fastqc (переменная называется before с расчётом на возможность тримминга). |
| hisat2 --no-softclip -x $idx -U $before -S $samfile 2> ${wdir}/map/out.txt | Картируем чтения. Так как сплайсинг возможен (картируем РНК на геном), убираем --no-spliced-alignment. Вывод сохраняем. |
| samtools view -b $samfile -o $bamfile | Получаем файл .bam. |
| samtools sort -o $sortedbam -O bam -T ./'sorttemp' $bamfile | Сортируем файл .bam, предоставляя samtools шаблон пути для временных файлов (-T). |
| samtools index $sortedbam | Индексируем сортированный файл .bam. |
| htseq-count -f bam -r pos -s no -i gene_id -m union $sortedbam $gtf > counting.o 2> counting.e | Подсчитываем чтения. Опции: -f bam для формата файла, -r pos для величины, по которой он отсортирован, -s no, чтобы не учитывать цепочку, -i gene_id для определения, чем будем индексировать/нумеровать референс, -m union — стандартный способ подсчёта перекрываний. Вывод сохраняем в файлы. |
| htseq-count -f bam -r pos -s no -i gene_id -m intersection-strict $sortedbam $gtf > counting_alt.o 2> counting_alt.e | Альтернативный подсчёт: считаем пересечение, только если чтение целиком легло на ген. (Дополнительное задание). |
| grep -vE '0$' counting.o | Оставляем только строки, не кончающиеся на 0, и узнвём, на какие гены ложатся чтения. |
| grep -vE '0$' counting_alt.o | Аналогично со вторым подсчётом. |
| grep ENSG00000167658.11 /nfs/srv/databases/ngs/Human/rnaseq_reads/*.gtf | less | Узнаём имя первого гена. (EEF2) |
| grep ENSG00000206775.1 /nfs/srv/databases/ngs/Human/rnaseq_reads/*.gtf | less | И второго. (SNORD37, это snoRNA — малая ядрышковая РНК). |
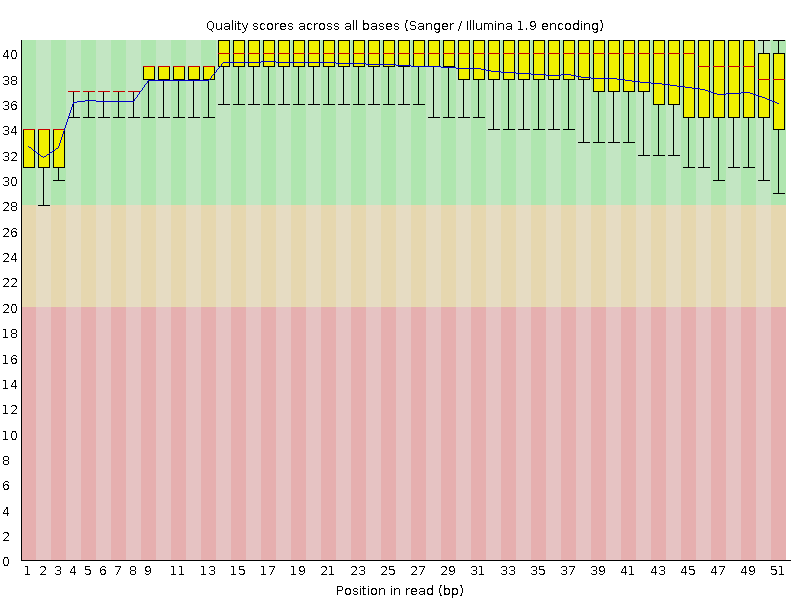
Первым шагом была проверка качества чтений (отчёт FastQC). Качество оказалось хорошим (см. рис. 1), только распределение нуклеотидов по частотам неестественное (объяснение — малая выборка генов, на которые ложатся риды; также при подборе случайных праймеров начало прочтения получается не вполне случайным: статья). Количество чтений равно 34936.
Смотрим вывод hisat2 и видим, что картировано 98,53 % чтений. Приведём также результаты работы grep для стандартного и альтернативного подсчёта. (См. нумерованный список ниже).
ENSG00000167658.11 34124 ENSG00000206775.1 6 __no_feature 284 __ambiguous 7 __not_aligned 515
ENSG00000167658.11 33858 ENSG00000206775.1 2 __no_feature 561 __not_aligned 515
Число вовсе не картированных ридов неизменно (и фиксировано после вызова hisat2), а вот число чтений, лёгших на гены, ожидаемо понизилось при вызове более строгого подсчёта. И всё-таки гены остались теми же. Куда попали не лёгшие в анотированные регионы чтения, неизвестно. Скорее всего, на интроны нашего гена, поскольку, например, ген snoRNA по координатам попал между экзонами основного гена.
Попавшийся мне ген EEF2 кодирует фактор элонгации 2 эукариот и играет роль в трансляции. SNORD37 кодирует snoRNA типа C/D, участвующую в 2'-O–метилировании рРНК.
После выполнения задания 11 к 12-му подступиться было проще.