В данном практикуме сравниваются методы спектроскопии ядерного магнитного резонанса и рентгеноструктурного анализа. Для этой цели мне выданы ID двух структур PDB, полученных этими методами. Мне достались 6J4I (ЯМР) и 1TGZ (РСА). В структуре ЯМР приводится 20 конформаций, разрешение X-ray структуры составляет $2{,}80$ Å.
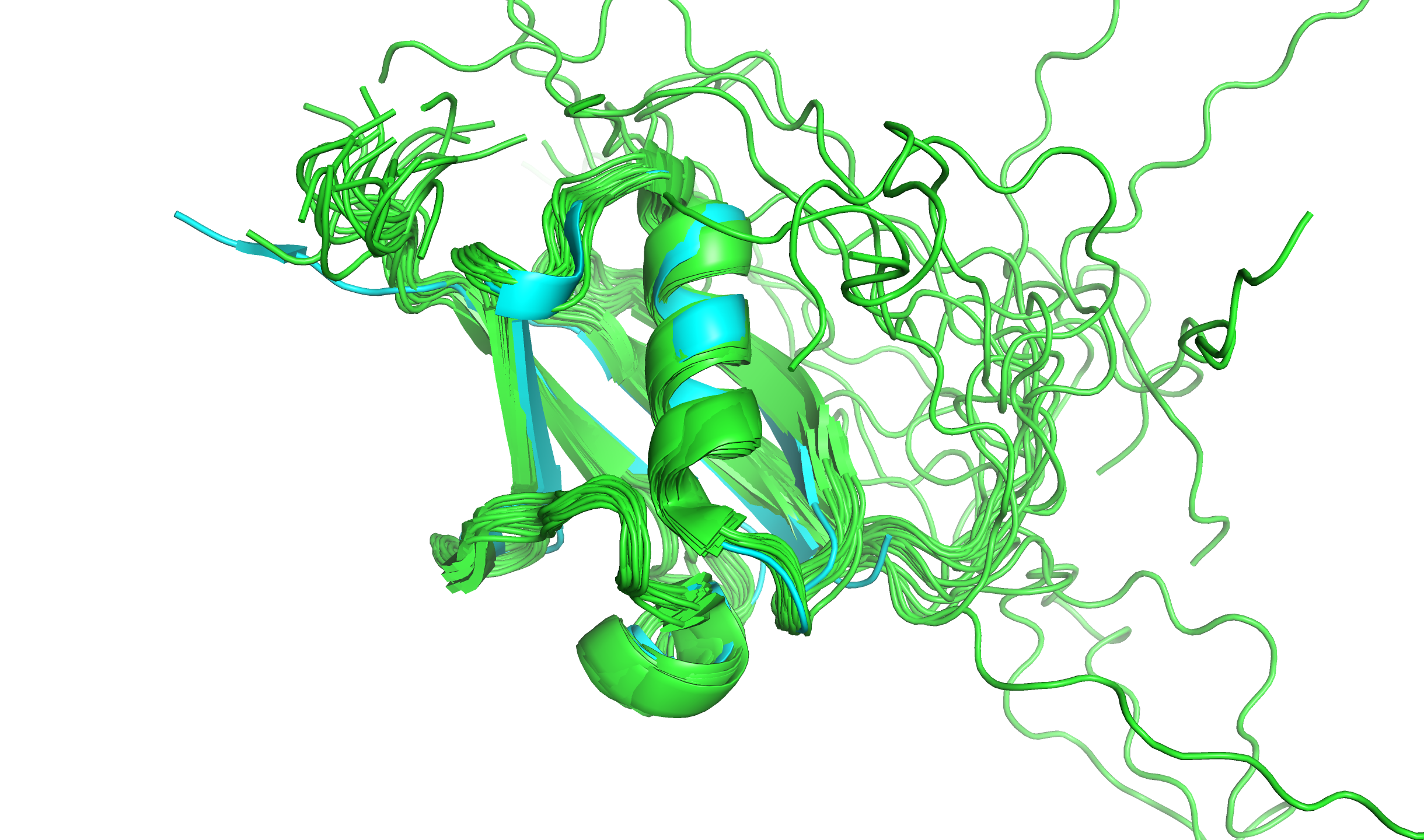
На рис. 1 изображены структуры в наложенном виде, ансамбль структур ЯМР показан зелёным цветом, модель РСА — бирюзовым.
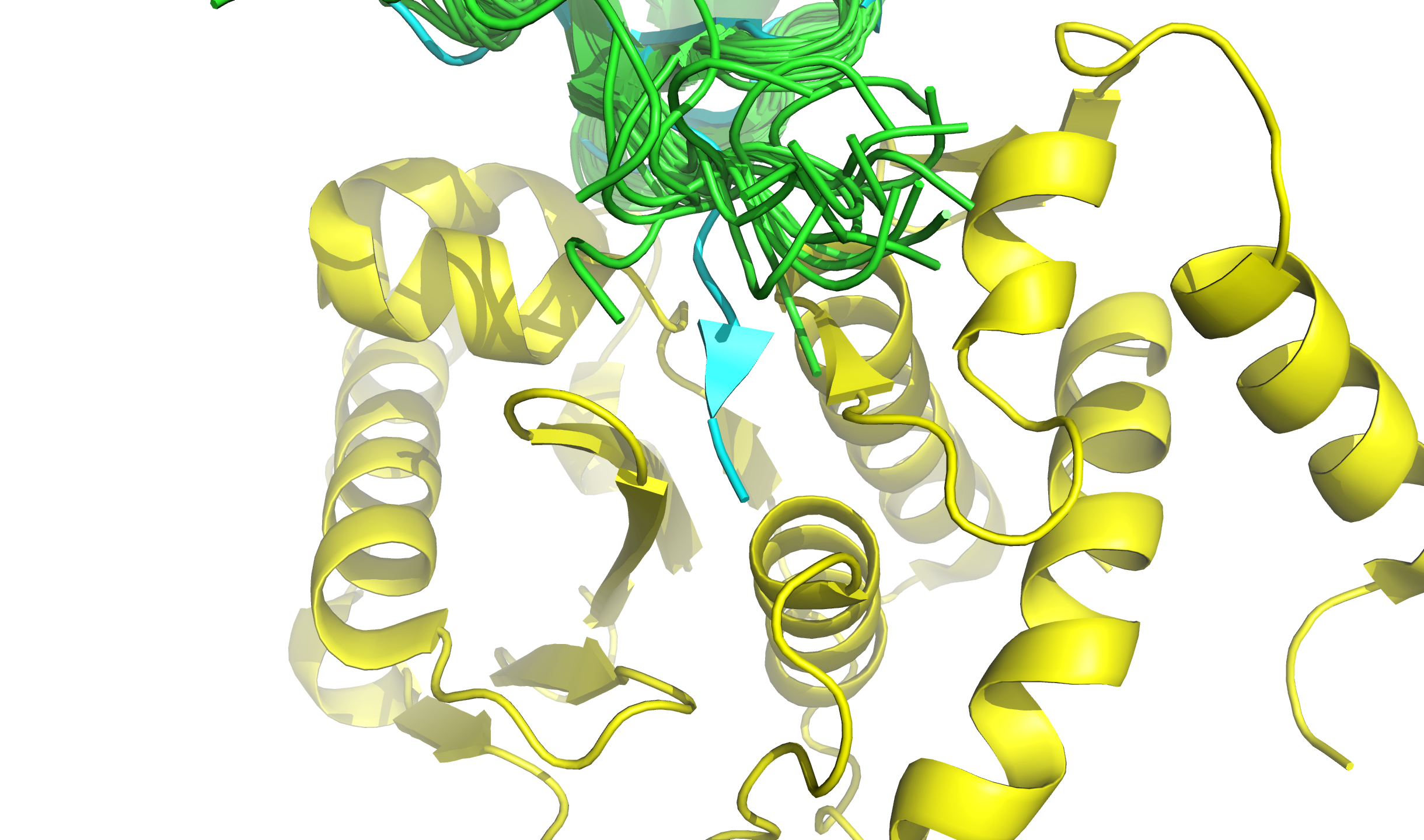
Можно отметить, что главные вторичные структуры белка, альфа-спирали и бета-листы ядра, в структуре РСА лежат в той же области, что и соответствующие элементы в структуре ЯМР — здесь наблюдается хорошее соответствие. Глобальные расхождения наблюдаем в терминальных областях: NTD в модели ЯМР имеет очень большой разброс между 20 конформациями, в структуре РСА же этот участок вовсе отсутствует. Либо NTD не разрешился, либо кристаллизацию проводили без этого участка. CTD же у структур более схожи, но в случае РСА аннотирован участок вторичной структуры. Это связано с тем, что кристаллизацию проводили в комплексе со вторым белком, и CTD нашей молекулы (SUMO-1) с ним взаимодействует (см. рис. 2).
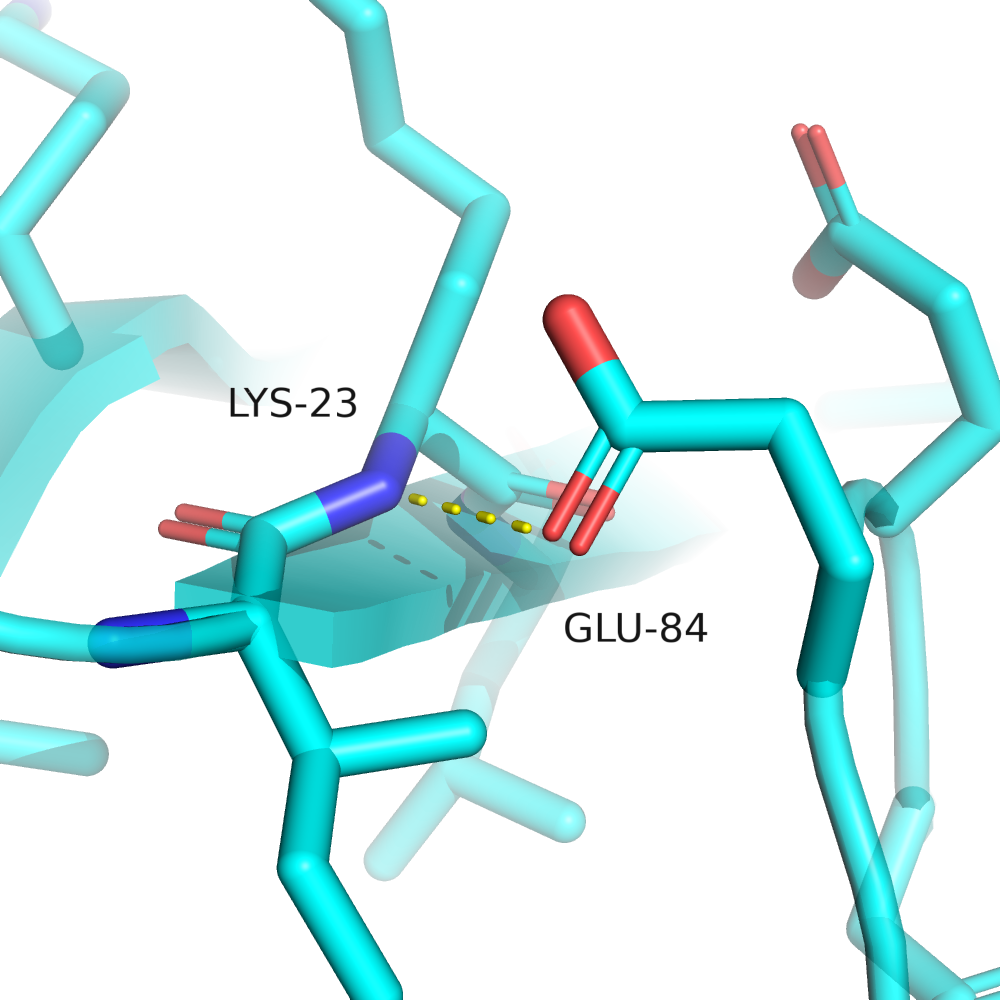
На микроуровне есть небольшие отличия в положении некоторых неструктурированных петель. Видимо, там образуются довольно слабые водородные связи, которые можно наблюдать в кристалле, но не в растворе (см. рис. 3).

Посмотрим, в какой степени RMSF, мера сходимости конформаций ансамбля ЯМР, может быть использована для интерпретации подвижности остатков и соответствует B-факторам из структуры РСА. Для этого построим график, сопоставив среднее RMSF и средний B-фактор для каждого из остатков, общих для двух структур.
import prody as pd, numpy as np
Загружаем структуру ЯМР.
nmr_model = pd.parsePDB("6J4I")
@> PDB file is found in working directory (6j4i.pdb.gz). @> 1543 atoms and 20 coordinate set(s) were parsed in 0.10s.
Считаем RMSF. Используем срез (islice), необходимые координаты подбираем по выравниванию в PyMol.
from itertools import islice
RMSFs = [np.mean(pd.calcRMSF(res)) for res in islice(nmr_model.iterResidues(), 22, 97)]
Аналогично поступим для РСА и B-факторов.
xray_model = pd.parsePDB("1TGZ")
@> PDB file is found in working directory (1tgz.pdb.gz). @> 2608 atoms and 1 coordinate set(s) were parsed in 0.04s.
mean_betas = [np.mean(res.getBetas()) for res in islice(xray_model["B"].iterResidues(), 3, 78)]
Построим график.
import matplotlib.pyplot as plt
%matplotlib inline
plt.plot(RMSFs, mean_betas, linestyle=" ", marker="o")
plt.title("Зависимость среднего B-фактора остатка \nв структуре РСА от RMSF в модели ЯМР", fontsize=18)
plt.xlabel("RMSF", fontsize=16)
plt.ylabel("B-фактор", fontsize=16)
plt.show()

Видно 4 выбивающиеся точки. Попробуем построить график без них.
plt.plot(RMSFs[:-4], mean_betas[:-4], linestyle=" ", marker="o")
plt.title("Зависимость среднего B-фактора остатка \nв структуре РСА от RMSF в модели ЯМР", fontsize=18)
plt.xlabel("RMSF", fontsize=16)
plt.ylabel("B-фактор", fontsize=16)
plt.show()

Так как понадобилось убрать 4 точки в конце массивов, они соответствуют CTD. У него большое RMSF и маленький B-фактор, что неудивительно, учитывая различие CTD, на которое я обратил внимание при общем сравнении структур, полученных двумя методами. У CTD действительно ограничена подвижность в кристалле по сравнению с раствором.
В целом тренда в точках не наблюдается, разве что можно сказать, что точки лежат в среднем выше диагонали, проведённой из левого нижнего в правый верхний угол. Иными словами, чаще у остатков с высоким B-фактором может быть низкий RMSF, чем наоборот, у остатков с высоким RMSF низкий B-фактор. Почему так происходит, мне непонятно.
Так как чёткого тренда нет, можно сказать, что зависимость у RMSF с B-фактором в нашем случае довольно слабая. Это может быть следствием как неполного соответствия структур (кейс CTD), так и большего влияния неполноты данных на RMSF, чем подвижности.
Для надёжности перепроверим, что на графики попали одни и те же остатки.
print(*islice(xray_model["B"].iterResidues(), 3, 78))
LYS 23 LEU 24 LYS 25 VAL 26 ILE 27 GLY 28 GLN 29 ASP 30 SER 31 SER 32 GLU 33 ILE 34 HIS 35 PHE 36 LYS 37 VAL 38 LYS 39 MET 40 THR 41 THR 42 HIS 43 LEU 44 LYS 45 LYS 46 LEU 47 LYS 48 GLU 49 SER 50 TYR 51 CYS 52 GLN 53 ARG 54 GLN 55 GLY 56 VAL 57 PRO 58 MET 59 ASN 60 SER 61 LEU 62 ARG 63 PHE 64 LEU 65 PHE 66 GLU 67 GLY 68 GLN 69 ARG 70 ILE 71 ALA 72 ASP 73 ASN 74 HIS 75 THR 76 PRO 77 LYS 78 GLU 79 LEU 80 GLY 81 MET 82 GLU 83 GLU 84 GLU 85 ASP 86 VAL 87 ILE 88 GLU 89 VAL 90 TYR 91 GLN 92 GLU 93 GLN 94 THR 95 GLY 96 GLY 97
print(*islice(nmr_model.iterResidues(), 22, 97))
LYS 23 LEU 24 LYS 25 VAL 26 ILE 27 GLY 28 GLN 29 ASP 30 SER 31 SER 32 GLU 33 ILE 34 HIS 35 PHE 36 LYS 37 VAL 38 LYS 39 MET 40 THR 41 THR 42 HIS 43 LEU 44 LYS 45 LYS 46 LEU 47 LYS 48 GLU 49 SER 50 TYR 51 CYS 52 GLN 53 ARG 54 GLN 55 GLY 56 VAL 57 PRO 58 MET 59 ASN 60 SER 61 LEU 62 ARG 63 LEU 64 LEU 65 PHE 66 GLU 67 GLY 68 GLN 69 ARG 70 ILE 71 ALA 72 ASP 73 ASN 74 HIS 75 THR 76 PRO 77 LYS 78 GLU 79 LEU 80 GLY 81 MET 82 GLU 83 GLU 84 GLU 85 ASP 86 VAL 87 ILE 88 GLU 89 VAL 90 TYR 91 GLN 92 GLU 93 GLN 94 THR 95 GLY 96 GLY 97
Да, всё хорошо.
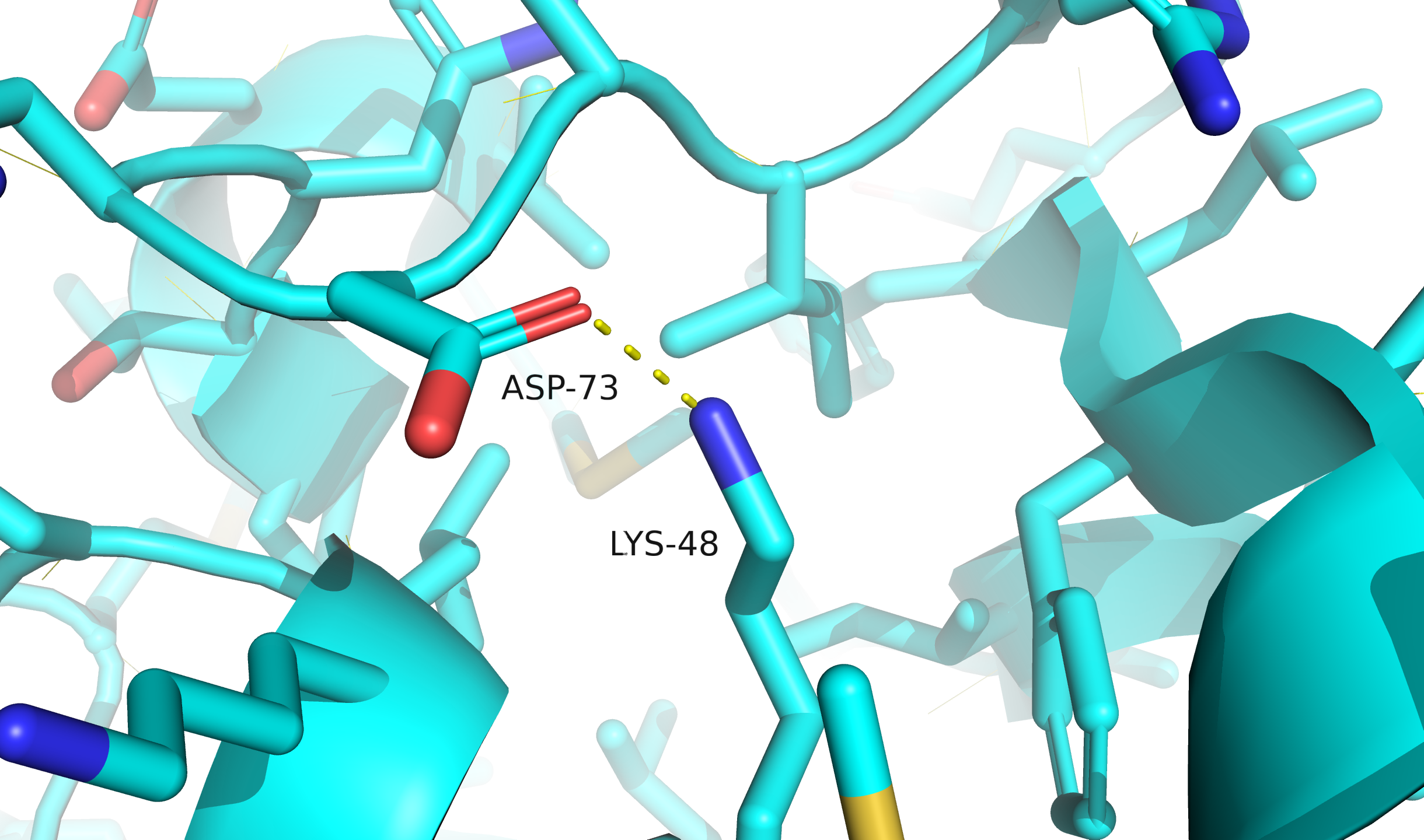
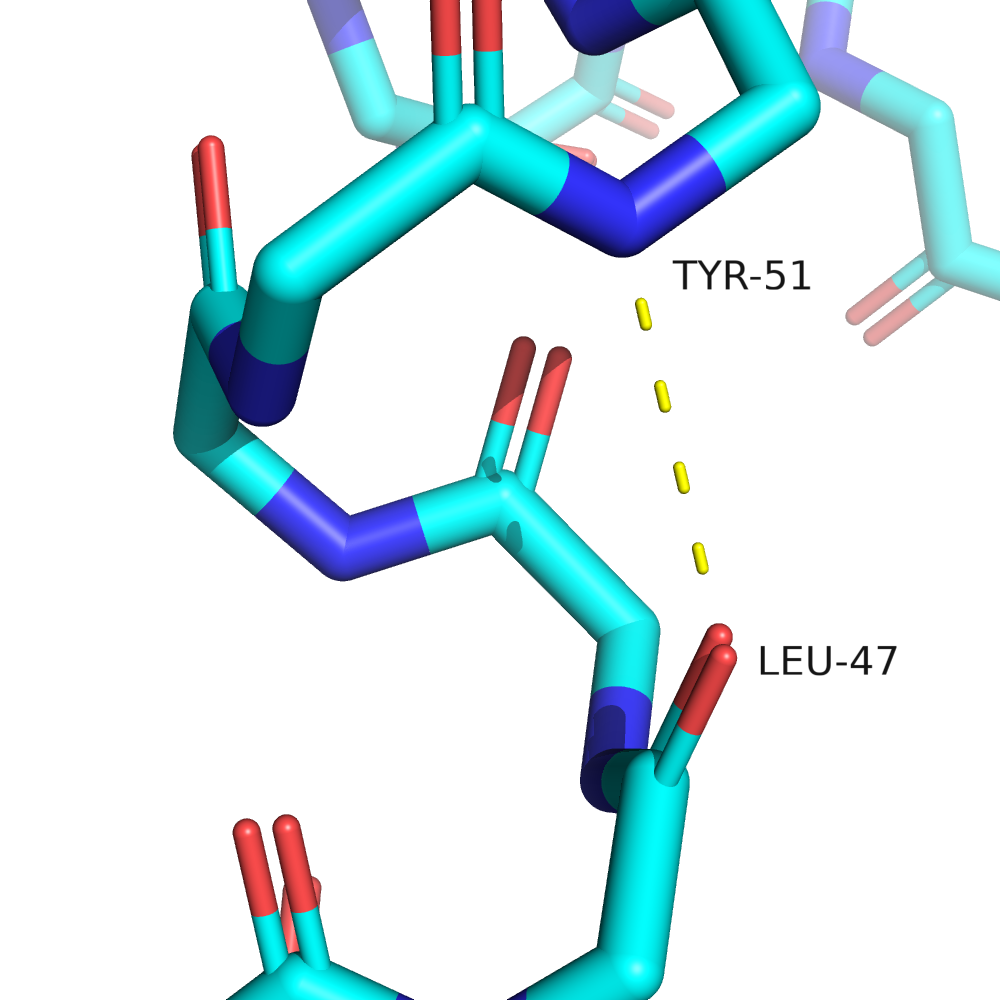
Выберем три водородные связи в РСА-структуре: в ядре белка между боковыми цепями аминокислотных остатков, в ядре белка между атомами остова, связь между атомами в экспонированной петле. Они изображены на рис. 4.
A.
Б.
В.
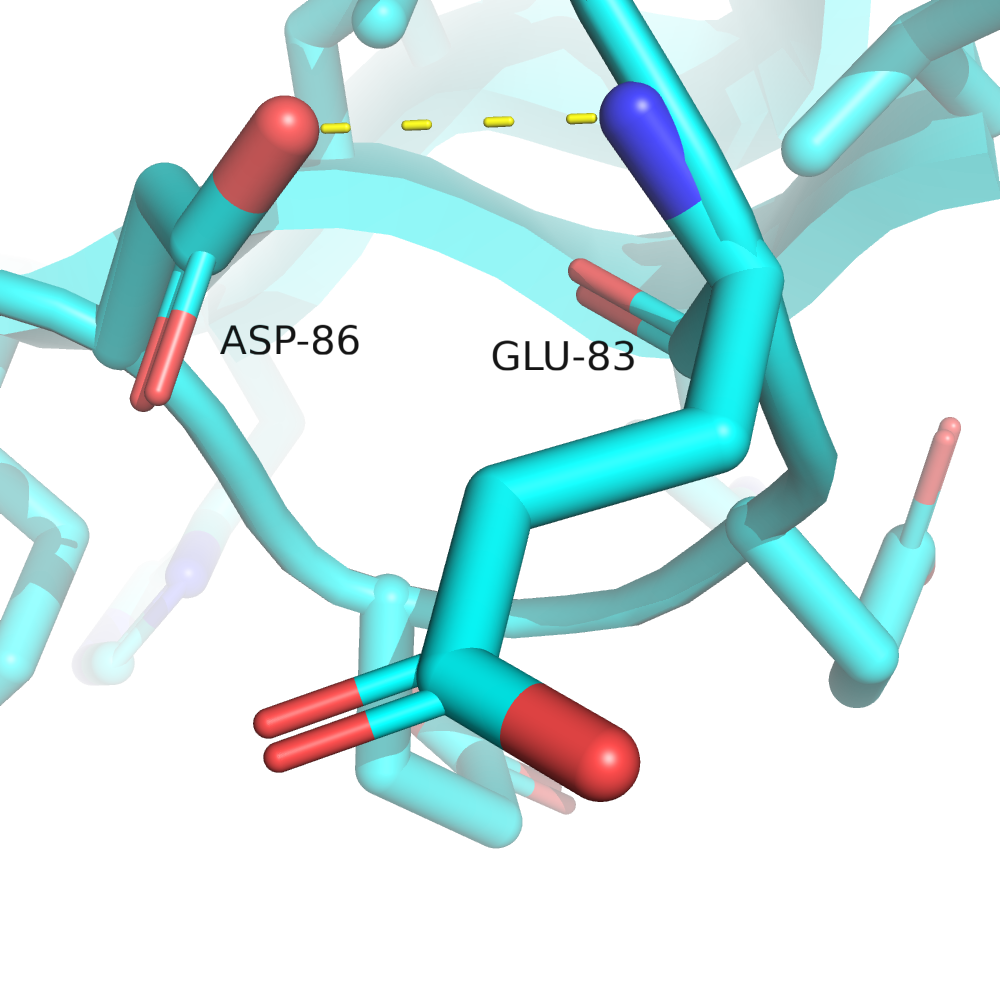
В первом случае связь между атомами $O_\delta$ D73 и $N_\varepsilon$ K48 (являющаяся и солевым мостиком). Так как белок довольно маленький, то эта связь всё равно не полностью изолирована от растворителя, но это лучшее, что я нашёл. 48-й лизин принадлежит к альфа-спирали. Вторая связь между атомами остова, $O$ из L47, $N$ из Y51, принадлежащих одной альфа-спирали. Третья связь между $O_\delta$ D86 и $N$ остова E83 в экспонированной петле.
Измерим расстояния во всех трёх случаях и запишем в numpy.array.
dist_int = np.array([7.1, 2.8, 4.2, 2.7, 3.0,
6.6, 6.1, 2.7, 4.6, 2.7,
2.7, 3.0, 6.1, 4.9, 5.4,
2.8, 2.8, 4.3, 2.7, 2.8])
dist_helix = np.array([3.3, 3.1, 3.3, 3.1, 3.0,
3.0, 3.1, 3.4, 3.7, 3.4,
3.4, 3.3, 2.9, 3.3, 2.9,
3.2, 3.3, 3.2, 3.1, 3.5])
dist_out = np.array([5.2, 4.5, 5.5, 5.6, 5.1,
5.0, 4.7, 5.4, 5.1, 4.7,
5.0, 5.1, 4.4, 4.5, 5.0,
4.9, 5.3, 4.1, 5.0, 5.3])
Таблица 1 характеризует выбранные связи в РСА- и ЯМР-модели.
| # | Расстояние РСА, Å | Число моделей ЯМР, где есть связь | % моделей ЯМР, где есть связь | Минимальное расстояние ЯМР, Å | Медианное расстояние ЯМР, Å | Максимальное расстояние ЯМР, Å |
|---|---|---|---|---|---|---|
| 1 | 2,8 | 11 | 55 | 2,7 | 3,0 | 7,1 |
| 2 | 3,1 | 19 | 95 | 2,9 | 3,25 | 3,7 |
| 3 | 2,6 | 0 | 0 | 4,1 | 5,0 | 5,6 |
Статистики были вычислены в numpy.
Итак, можно видеть, что водородные связи устойчиво сохраняются только в мотиве вторичной структуры (альфа-спирали). Здесь водородных связей много, они поддерживают друг друга, а также благоприятная конформация торсионных углов у $C_\alpha$-атомов. Водородная связь между боковыми радикалами погружённых в глобулу аминокислот наблюдается примерно в половине конформаций ансамбля ЯМР, возможно, это число было бы больше, если бы гидрофобное ядро окружало эти аминокислоты со всех сторон (с другой стороны, такое можно встретить довольно редко, ведь тогда у полярных аминокислот явно некомфортное окружение). Крайний случай, когда растворитель явно выигрывает конкуренцию за водородные связи, можно увидеть в третьем случае. Кстати, именно эта петля сильнее всего различалась в структурах РСА и ЯМР.