
При получении данных о структуре молекулы белка методом рентгеновской кристаллографии важным этапом является оптимизация первичной, черновой модели. Оптимизированная модель требует валидации, чему и посвящён этот практикум.
Интересно, что в оригинальной публикации исследуется именно влияние наличия остатка в запрещённой конформации на карте Рамачандрана на свойства белка в целом — получается, один из маргинальных в этом смысле остатков введён в структуру искуственно, заменой G25A. Авторы узнали, что в данном случае остаток аланина принимает запрещённую конформацию с минимальным изменением третичной структуры, но возникшее локальное напряжение значительно снижает стабильность белка.
Изучим отчёт о валидации структуры. В нём приведены такие метрики, как $\mathrm R_{free}$, clashscore (количество необычно близких атомов на 1000), доля маргинальных остатков на карте Рамачандрана, а ещё — маргинальных остатков по конформации бокового радикала и, наконец, маргинальных остатков по $z$-статистике пространственного R-фактора. Приведём их значения в таблице 1.
| Параметр | Значение |
|---|---|
| $\mathrm R_{free}$ | 0,200 |
| Clashscore | 3 |
| Ramachandran outliers | 0,5 % |
| Sidechain outliers | 1,6 % |
| RSRZ outliers | 0 % |
Единственный остаток в запрещённой зоне на карте Рамачандрана (из 213), который фактически и исследуется, даёт долю маргинальных остатков в 0,5 %, что выше, чем у примерно 90 % структур со сходным разрешением (номинальное разрешение изучаемой структуры 1,64 Å). Однако у остова всех остальных остатков конформация в норме. Есть три остатка, маргинальных по конформации боковой цепи (у примерно 60 % структур сходного разрешения ситуация лучше), на них мы обратим внимание позже. Остальные параметры структуры скорее хорошие, диаграмма с перцентилями для всех структур РСА и структур похожего разрешения из валидационного отчёта приведена на рис. 1.
Также отчёт показывает, что в расшифровке структуры нет участков, которые бы плохо соответствовали экспериментальной электронной плотности. (The upper red bar (where present) indicates the fraction of residues that have poor fit to the electron density. — Красных черт на соответствующей диаграмме нет.)
Теперь для оценки качества расшифровки структуры обратимся к MolProbity. Загрузим структуру и проведём сначала анализ геометрии, не добавляя водороды. Результат приведён в таблице 2.
| Protein Geometry |
Poor rotamers | 3 | 1.60% | Goal: <0.3% |
| Favored rotamers | 181 | 96.79% | Goal: >98% | |
| Ramachandran outliers | 1 | 0.47% | Goal: <0.05% | |
| Ramachandran favored | 205 | 97.16% | Goal: >98% | |
| Rama distribution Z-score | -0.71 ± 0.52 | Goal: abs(Z score) < 2 | ||
| Cβ deviations >0.25Å | 3 | 1.48% | Goal: 0 | |
| Bad bonds: | 7 / 1726 | 0.41% | Goal: 0% | |
| Bad angles: | 5 / 2336 | 0.21% | Goal: <0.1% | |
| Peptide Omegas | Cis Prolines: | 1 / 6 | 16.67% | Expected: ≤1 per chain, or ≤5% |
| Additional validations | Chiral volume outliers | 0/277 | ||
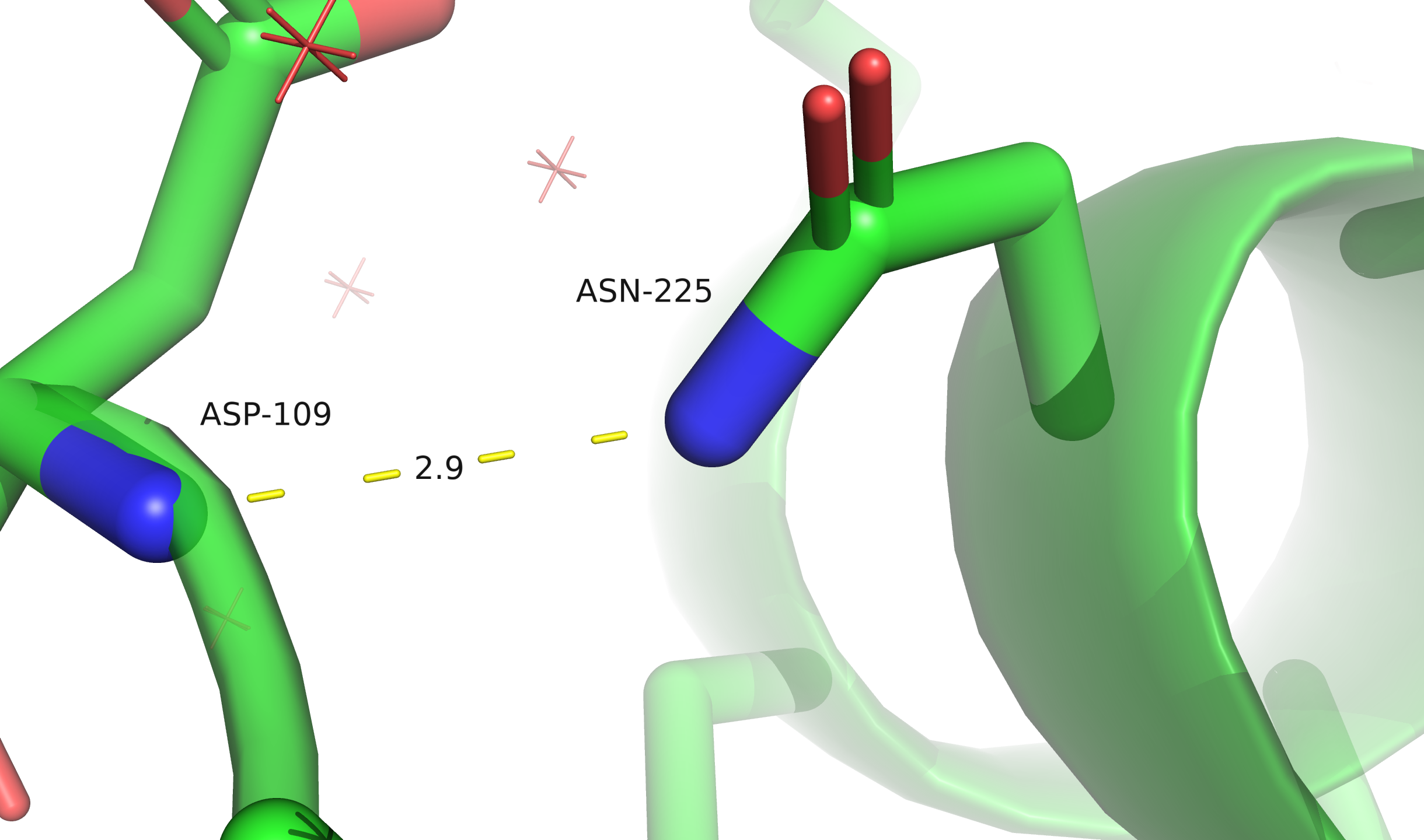
Так же, как и в отчёте wwPDB, присутствует 3 маргинальных ротамера, 1 аутлаер на карте Рамачандрана, но MolProbity в дополнение к этому оценивает и отклоняющиеся значения длин ковалентных связей и углов между связями. Изучим отчёт подробнее (откроем Multi-criterion chart). Интересно, что набор плохих ротамеров здесь и в отчёте wwPDB не совпал: в валидационном отчёте это были остатки S26, R64, D183, а в выдаче MolProbity D183 в допустимой области, зато проблемы с E42. Эти 4 маргинальных ротамера приведены на рис. 2.


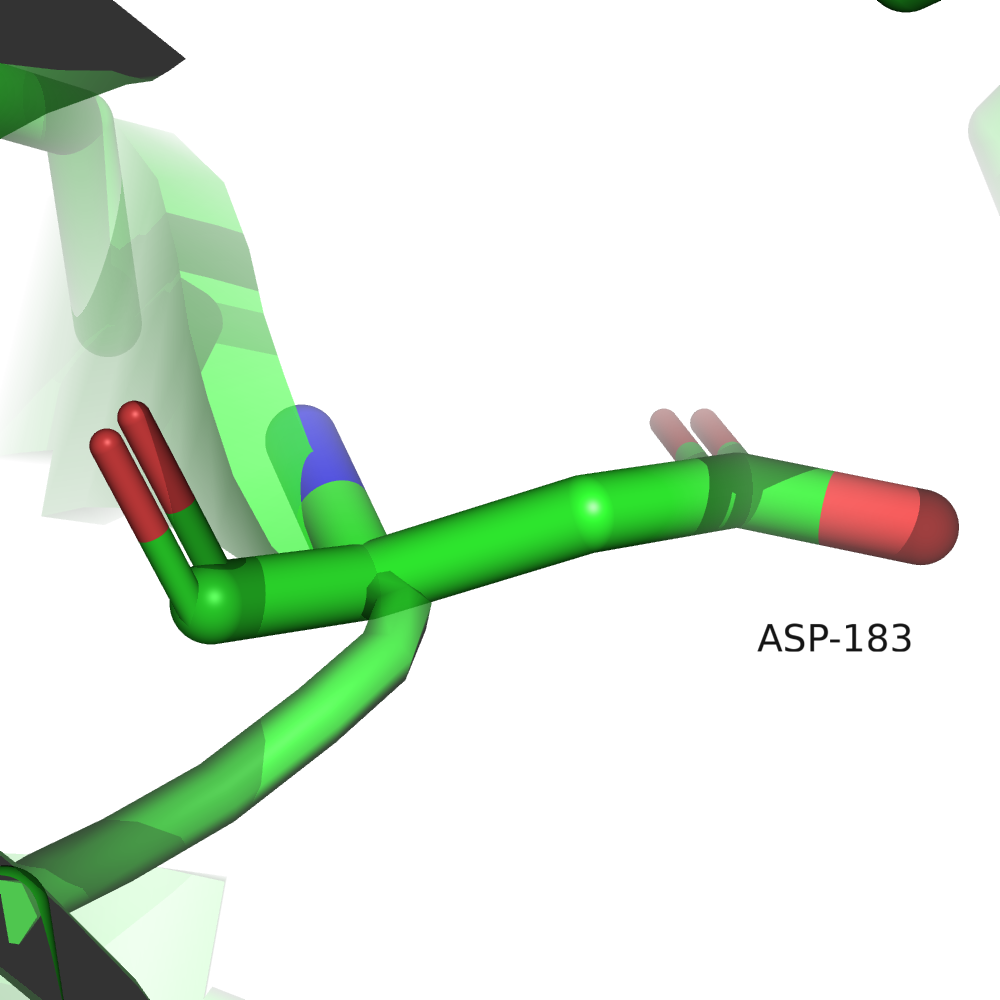
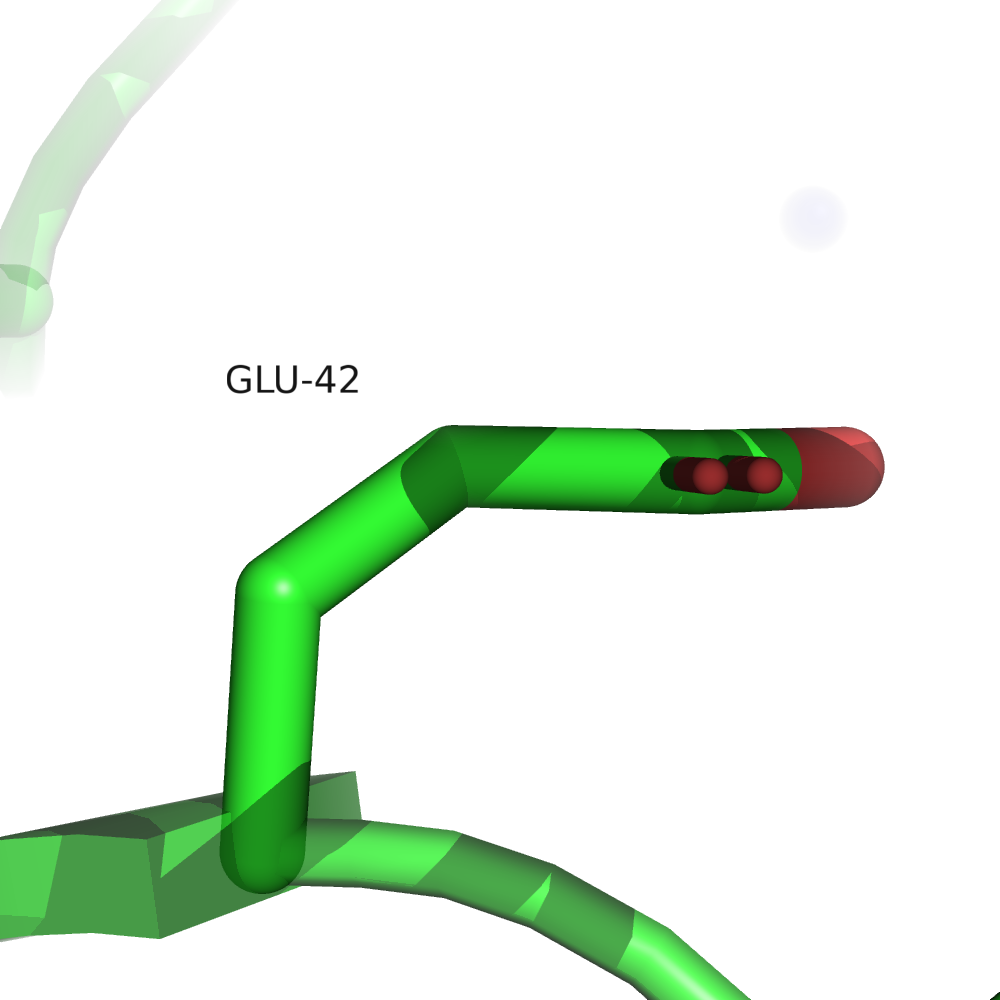
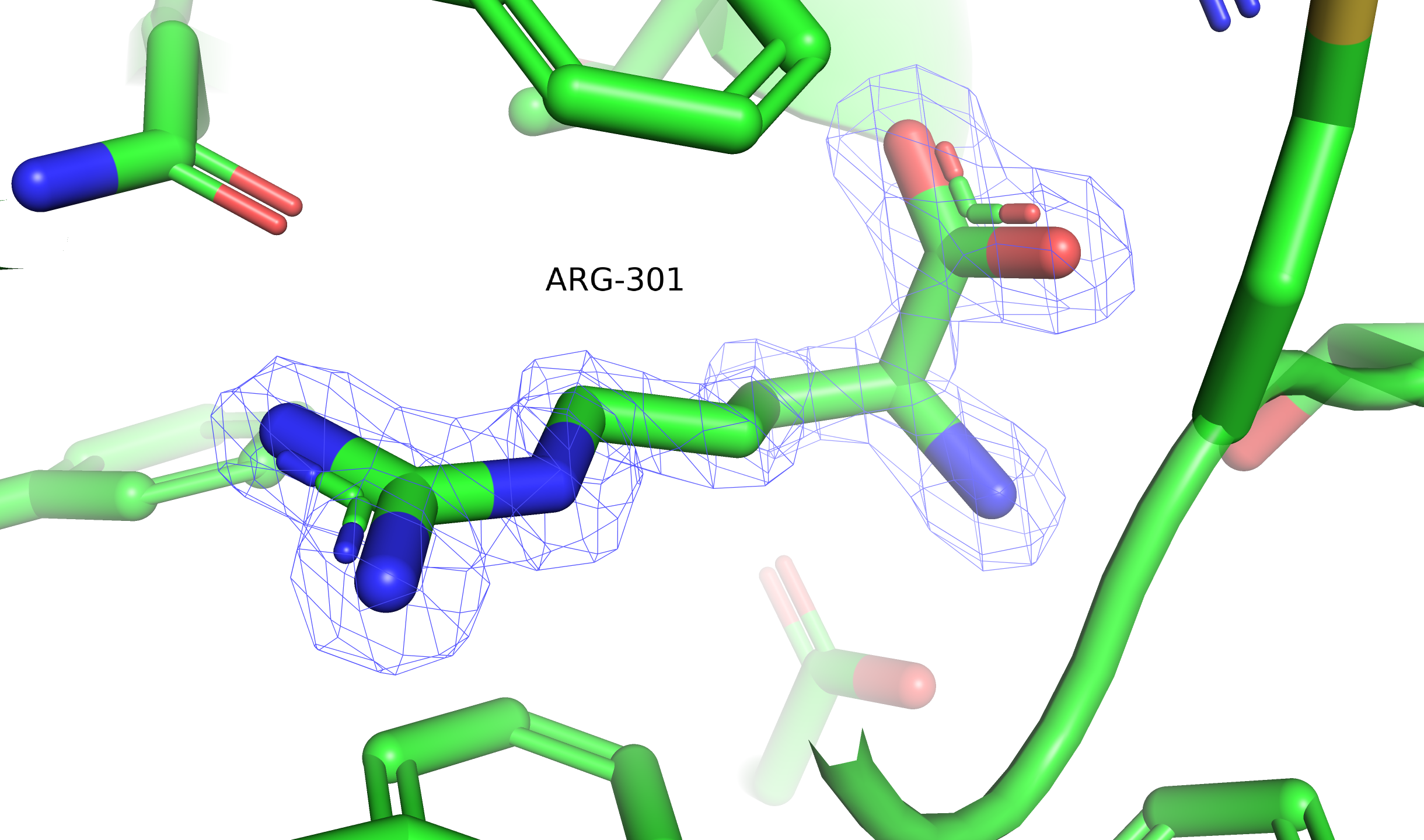
Ser26 находится в конформации, близкой к заслонённой, Glu42 и Arg64 необычно изогнуты и прижаты к остову, у Asp183 необычно сближены кислород карбоксильной группировки и NH-группа остова.
Также MolProbity даёт примеры маргинальных остатков с точки зрения длин ковалентных связей и углов между ними — обратим внимание на такие остатки (примеры приведены на рис. 3).
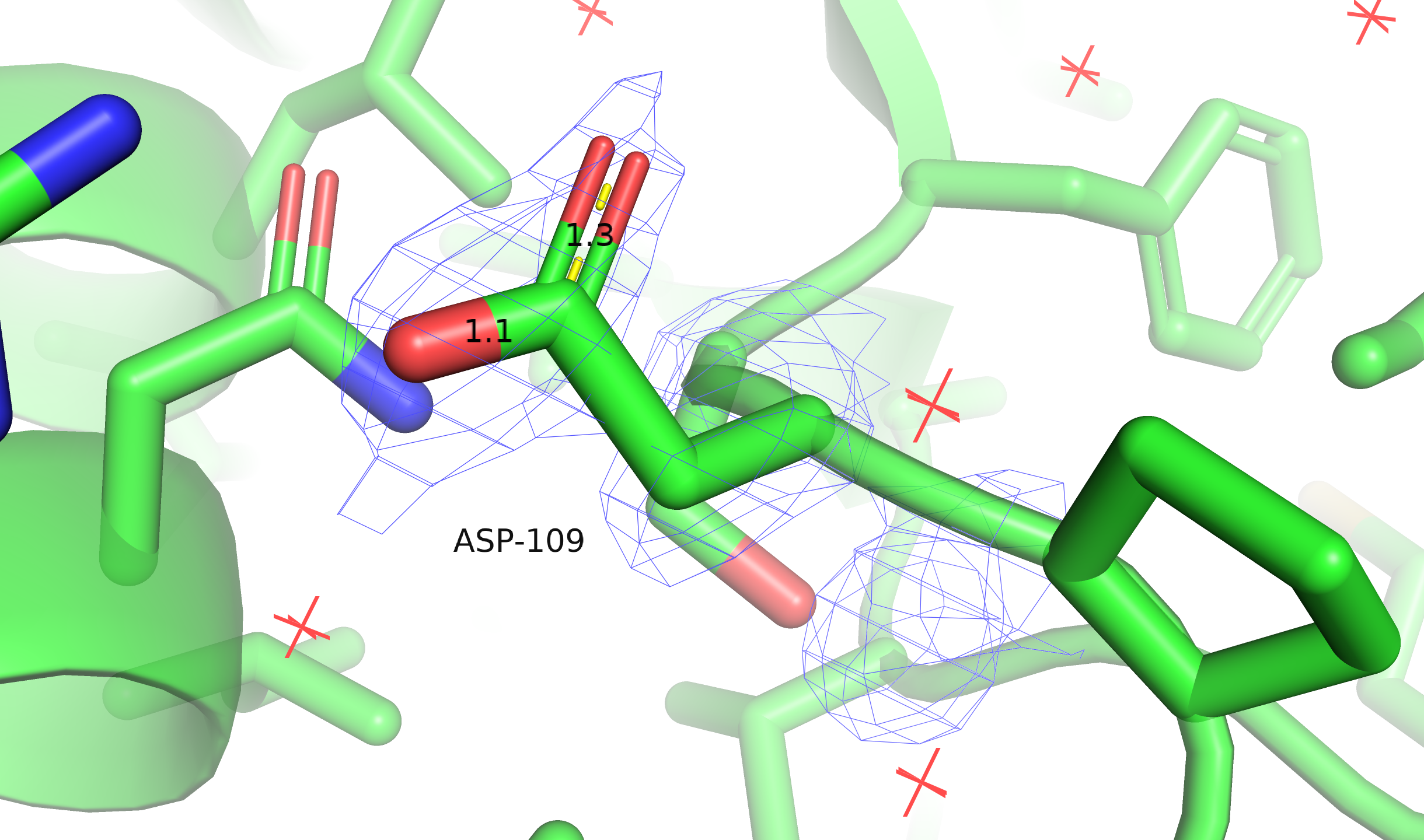
На левой половине рисунка одна из связей с кислородом в карбоксильной группе аномально укорочена, у остатка из правой части сразу две проблемы — аномально длинная связь и угол, далёкий от тетраэдрического. При этом в электронную плотность они как-то вписаны, и удовлетворительно, если верить отчёту wwPDB. Возможно, здесь источником шума в расшифровке функции плотности были молекулы воды (на рисунке они тоже показаны).
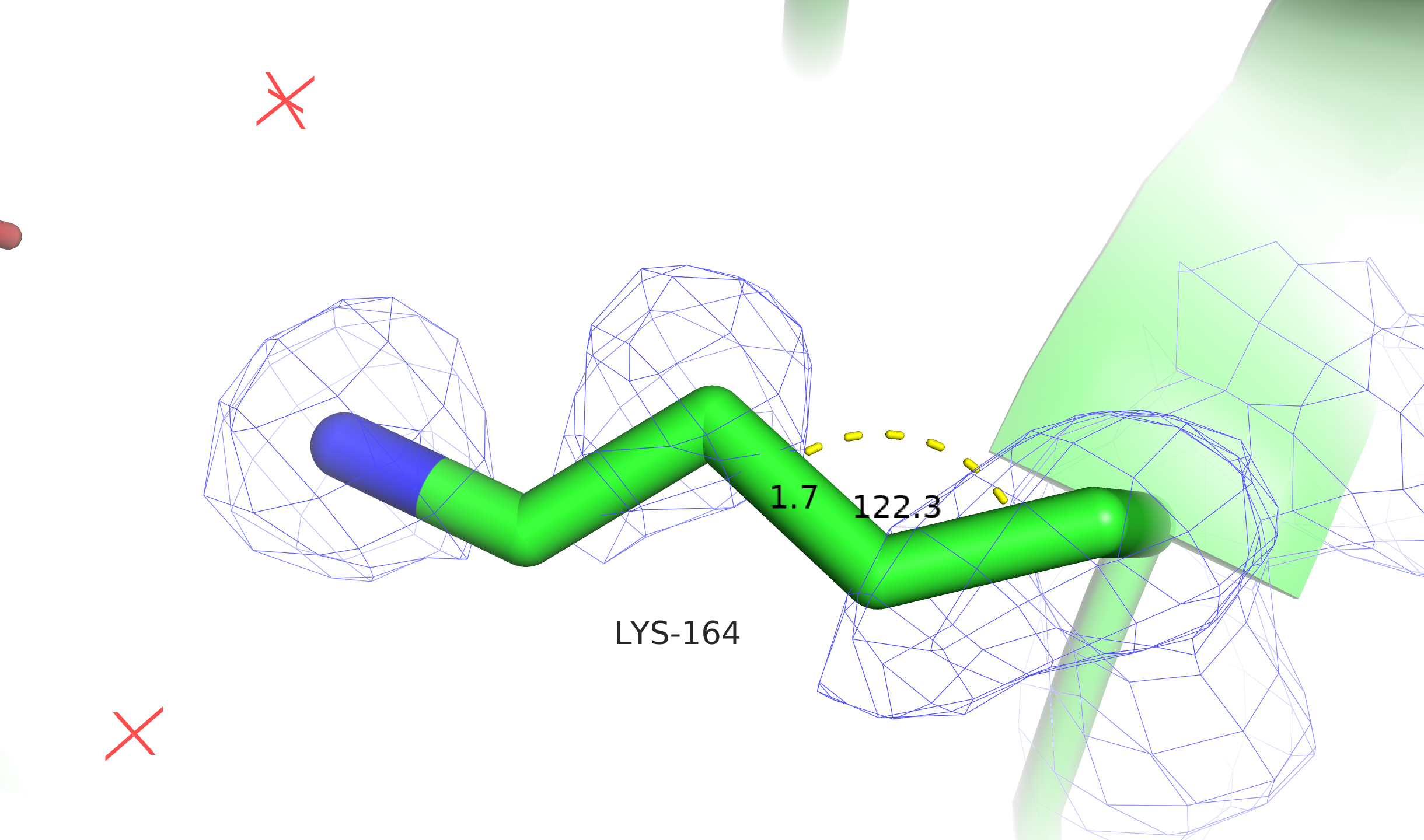
Добавим в структуру водороды. При этом MolProbity сообщает, что один из остатков требует разворота. Посмотрим на него (см. рис. 4).
Видно, что остаток действительно нужно развернуть: сейчас наблюдается близкий контакт $\mathsf{NH}_2$ и $\mathsf{NH}$-групп, а после разворота будет хорошая водородная связь.
После добавления водородов проверим геометрию снова — со всеми доступными контактами. Сводная таблица представлена ниже (табл. 3).
| All-Atom Contacts |
Clashscore, all atoms: | 2.62 | 99th percentile* (N=708, 1.64Å ± 0.25Å) | |
| Clashscore is the number of serious steric overlaps (> 0.4 Å) per 1000 atoms. | ||||
| Protein Geometry |
Poor rotamers | 3 | 1.60% | Goal: <0.3% |
| Favored rotamers | 181 | 96.79% | Goal: >98% | |
| Ramachandran outliers | 1 | 0.47% | Goal: <0.05% | |
| Ramachandran favored | 205 | 97.16% | Goal: >98% | |
| Rama distribution Z-score | -0.71 ± 0.52 | Goal: abs(Z score) < 2 | ||
| MolProbity score^ | 1.34 | 95th percentile* (N=7267, 1.64Å ± 0.25Å) | ||
| Cβ deviations >0.25Å | 3 | 1.48% | Goal: 0 | |
| Bad bonds: | 7 / 1726 | 0.41% | Goal: 0% | |
| Bad angles: | 5 / 2336 | 0.21% | Goal: <0.1% | |
| Peptide Omegas | Cis Prolines: | 1 / 6 | 16.67% | Expected: ≤1 per chain, or ≤5% |
| Additional validations | Chiral volume outliers | 0/277 | ||
| Waters with clashes | 15/272 | 5.51% | See UnDowser table for details | |
По сравнению с тем, что было раньше, здесь появились Clashscore, MolProbity score и проверка на пересечения с молекулами воды. Действительно, при поиске пересечений есть смысл учитывать наличие атомов водорода.
У изучаемой структуры довольно высокое разрешение, а все остатки в целом соответствуют экспериментальной электронной плотности. Наблюдаемый остаток в запрещённой области на карте Рамачандрана там и должен находиться, нас может смутить большое количество аномальных ротамеров. Попробуем приглядеться к ним (см. рис. 5).

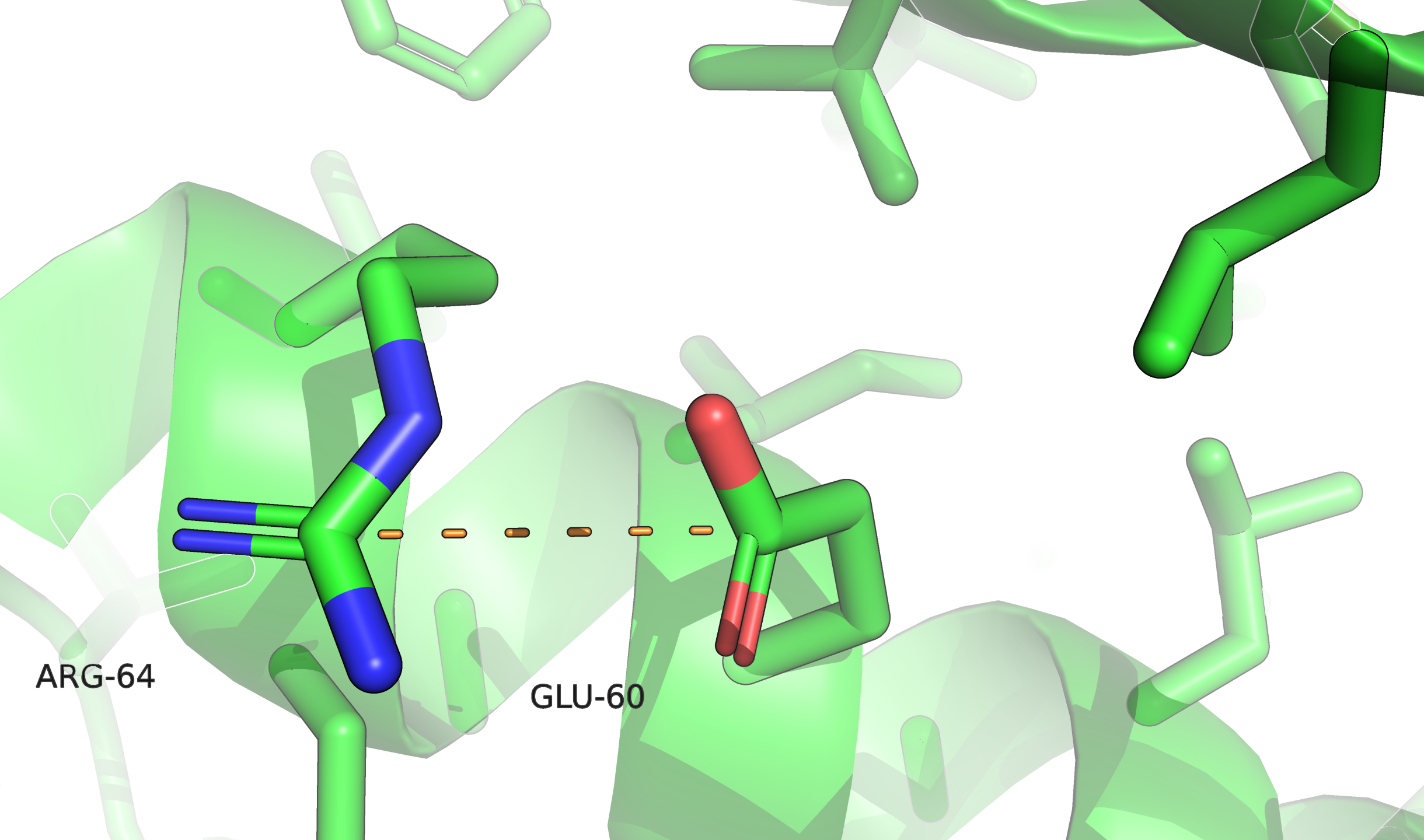
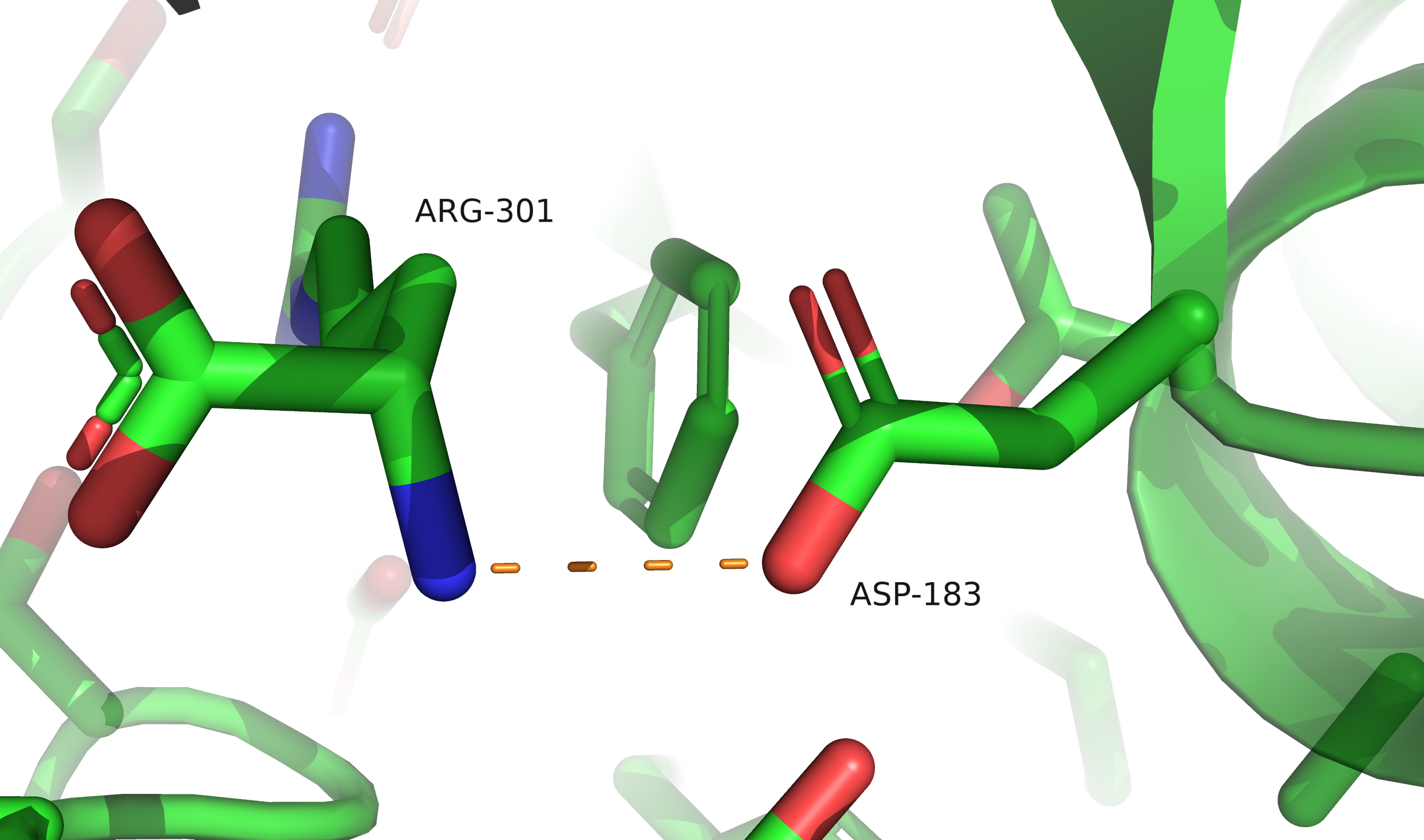
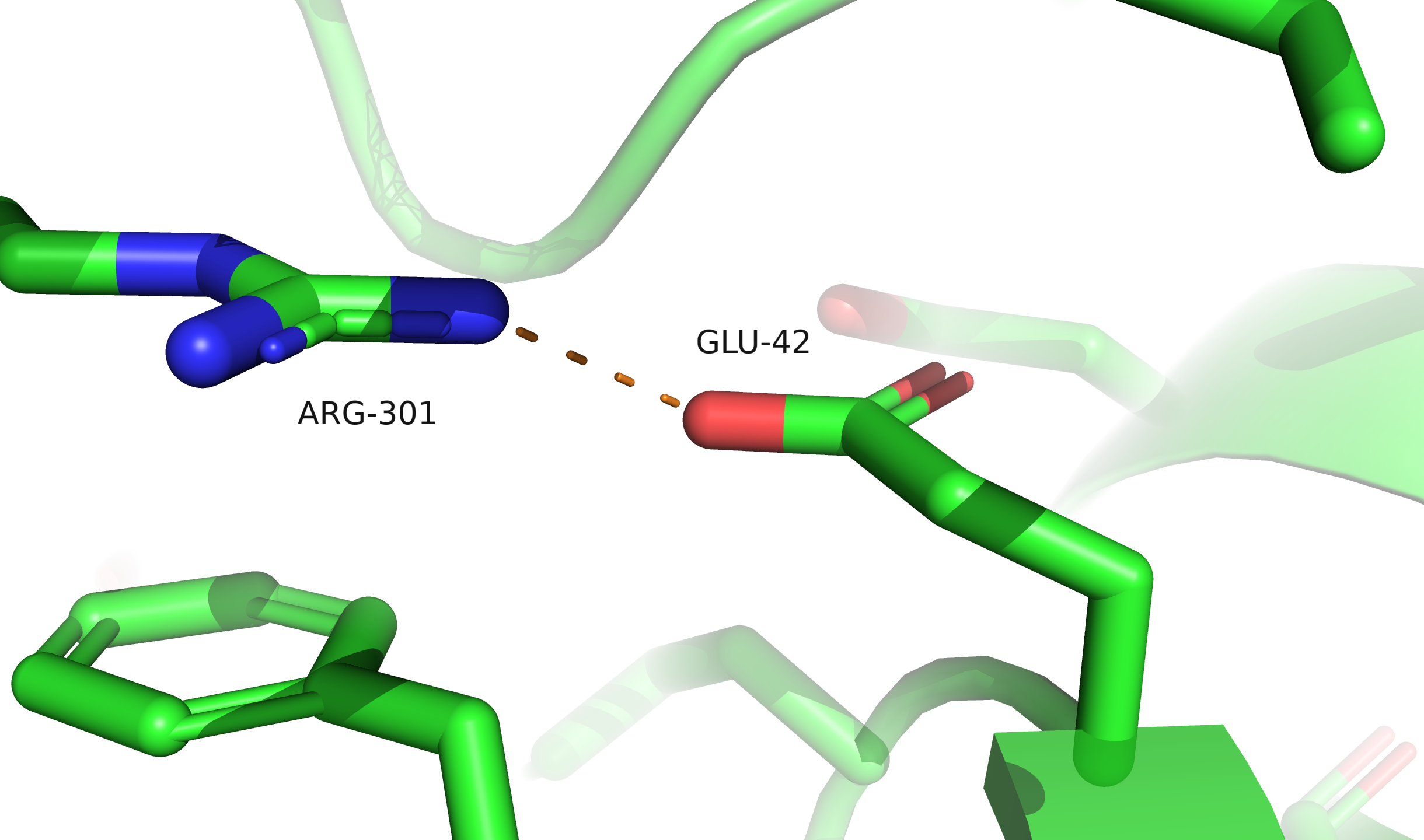
Ser26, по всей видимости, принимает заслонённую конформацию из-за близости соседней молекулы в кристалле (показано отображение surface). По-видимому, в функциональном состоянии белка этот остаток развёрнут не так. Остатки Asp183 и Glu42 образуют солевые мостики с лигандом — молекулой аргинина. Это однозначно функционально важные остатки. Остаток Arg64 образует солевой мостик с глутаматом из соседнего витка альфа-спирали, если такое взаимодействие наблюдается и в функциональном состоянии, возможно, оно дополнительно стабилизирует вторичную структуру, что особенно важно для термофильной бактерии.
Качество электронной плотности довольно хорошее, что можно ожидать при разрешении 1,64 Å и полноте 96,6 %. Только для нескольких остатков шум даёт аномалии в расшифровке, выражающиеся в нетипичных длинах ковалентных связей и углах между ними. Я бы сказал, что полученная расшифровка вполне пригодна для изучения особенностей исследуемого белка — и уж точно подходит для определения торсионных углов при $\mathrm{C}_\alpha$-атоме одного остатка.
Можно отметить, что есть электронная плотность и для лиганда, с этим всё в порядке (см. рис. 6).
Найдём наш PDB ID на сервисе PDB Redo. На странице можно оценить изменения разнообразных метрик, а ещё там показано, для каких остатков получено значимое изменение качества вписанности в электронную плотность. В нашем случае есть только одно значимое изменение — улучшение для Met74. Этот остаток показан на рис. 6.
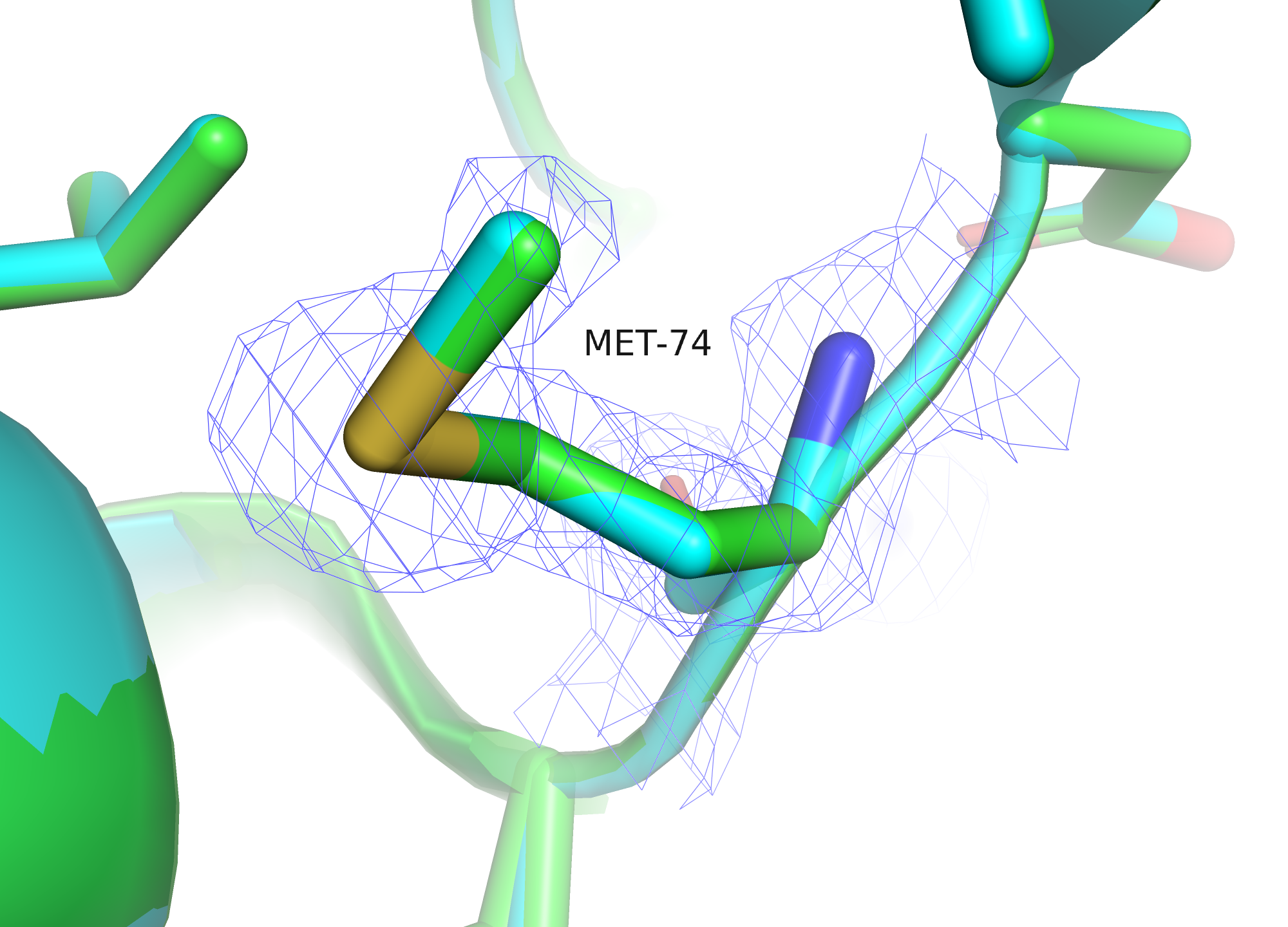
Здесь разве что чуть-чуть сдвинулся $C_\varepsilon$-атом, изменение почти незаметно глазу.
Для сравнения с моделью из RCSB PDB проведём аналогичные анализы в MolProbity — базовый анализ геометрии и анализ с учётом всех контактов. Сводные таблицы для этих запросов приведены ниже (табл. 4–5).
| Protein Geometry |
Poor rotamers | 2 | 1.07% | Goal: <0.3% |
| Favored rotamers | 180 | 96.26% | Goal: >98% | |
| Ramachandran outliers | 1 | 0.47% | Goal: <0.05% | |
| Ramachandran favored | 206 | 97.63% | Goal: >98% | |
| Rama distribution Z-score | -0.78 ± 0.50 | Goal: abs(Z score) < 2 | ||
| Cβ deviations >0.25Å | 0 | 0.00% | Goal: 0 | |
| Bad bonds: | 3 / 1726 | 0.17% | Goal: 0% | |
| Bad angles: | 13 / 2336 | 0.56% | Goal: <0.1% | |
| Peptide Omegas | Cis Prolines: | 1 / 6 | 16.67% | Expected: ≤1 per chain, or ≤5% |
| Additional validations | Chiral volume outliers | 0/277 | ||
| All-Atom Contacts |
Clashscore, all atoms: | 2.91 | 99th percentile* (N=708, 1.64Å ± 0.25Å) | |
| Clashscore is the number of serious steric overlaps (> 0.4 Å) per 1000 atoms. | ||||
| Protein Geometry |
Poor rotamers | 2 | 1.07% | Goal: <0.3% |
| Favored rotamers | 180 | 96.26% | Goal: >98% | |
| Ramachandran outliers | 1 | 0.47% | Goal: <0.05% | |
| Ramachandran favored | 206 | 97.63% | Goal: >98% | |
| Rama distribution Z-score | -0.78 ± 0.50 | Goal: abs(Z score) < 2 | ||
| MolProbity score^ | 1.24 | 98th percentile* (N=7267, 1.64Å ± 0.25Å) | ||
| Cβ deviations >0.25Å | 0 | 0.00% | Goal: 0 | |
| Bad bonds: | 3 / 1726 | 0.17% | Goal: 0% | |
| Bad angles: | 13 / 2336 | 0.56% | Goal: <0.1% | |
| Peptide Omegas | Cis Prolines: | 1 / 6 | 16.67% | Expected: ≤1 per chain, or ≤5% |
| Additional validations | Chiral volume outliers | 0/277 | ||
| Waters with clashes | 7/269 | 2.60% | See UnDowser table for details | |
Стало меньше плохих ротамеров и связей с аномальной длиной, зато увеличилось количество аномальных углов. Стало меньше пересечений с молекулами воды, хотя весь Clashscore чуть-чуть ухудшился. Квантиль MolProbity score выросла с 95 % до 98 % (небольшое улучшение).
Из плохих ротамеров остались Ser26 и Glu42. Arg64 в модели RCSB PDB был маргиналом по конформации боковой цепи и по длине одной из связей, теперь же у него допустимый ротамер и длины, но один из ковалентных углов нетипичен. Можно ещё отметить, что при добавлении водородов к модели PDB Redo не понадобились перевороты остатков Asn/Gln/His.
Подводя итог, можно сказать, что и структура из PDB Redo годится для изучения особенностей белка: принципиальной разницы с RCSB PDB я не вижу.