Для начала в директории nfs/srv/databases/ngs/ была создана папка s.kozyulina, куда были скопированы файлы chr17.fasta (файл с хромосомой) и chr17.fastq (файл с ридами).
Анализ качества и очистка чтений.
| № команды | Команда | Результат выполнения команды |
| 1 | fastqc chr17.fastq | анализ чтений. Выдача данной команды - файл chr17_fastqc.html |
| 2 | java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr17.fastq chr17clean.fastq TRAILING:20 MINLEN:50 | команда для очистки чтений: отрезает с конца каждого чтения нуклеотиды с качеством ниже 20 и оставляет только чтения длины не менее 50 нуклеоидов. |
| 3 | fastqc chr17clean.fastq | анализ очищенного прочтения. Полученный файл - chr17clean_fastqc.html |
| Характеристики | Исходные чтения | Чтения после чистки |
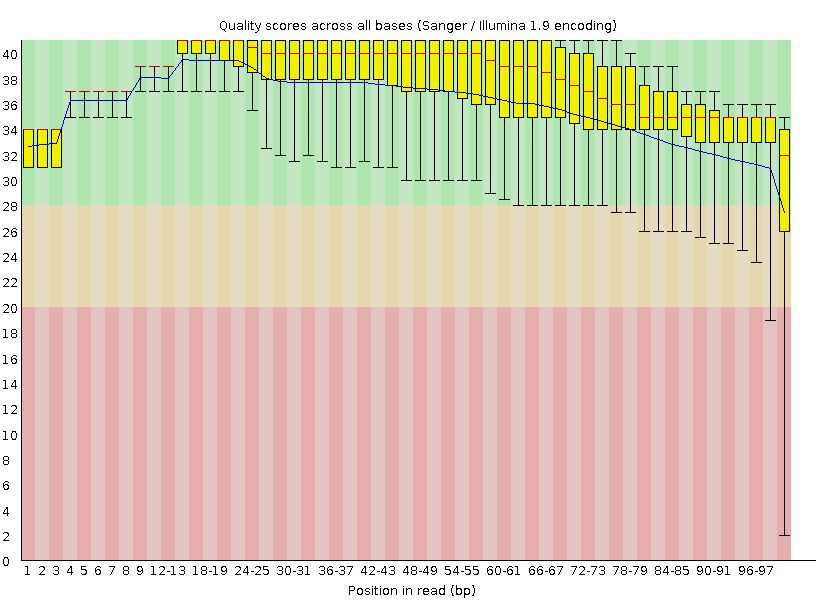
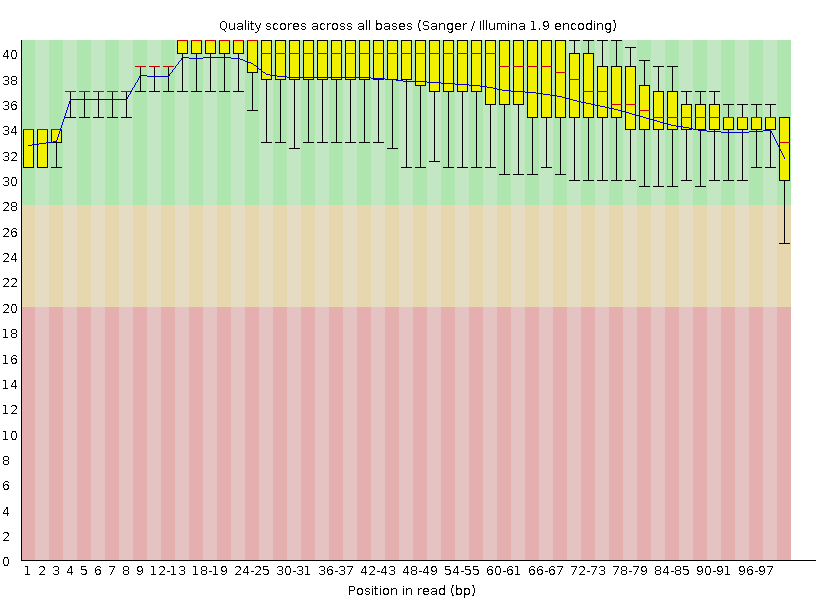
| График "Per base quality" из программы FastQC | |
|
| Число чтений | 11046 | 10868 |
| Что изменилось? | В результате чистки программой trimmomatic было удалено 178 чтений длиной меньше 50 нуклеотидов. В итоге остались только чтения длиной 50-100 нуклеотидов, в то время как длина исходных чтений варьировалась в промежутке 31-100 нуклеотидов. | |
Комментарии по чтению графиков "Per base quality":
Картирование чтений.
На данном этапе необходимо сначала проиндексировать референсную последовательность, а затем построить выравнивание прочтений и референса в формате .sam (ход действий представлен ниже).
| № команды | Команда | Результат выполнения команды |
| 1 | export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 | вызывает программу hisat2-2.0.5, лежащую в данной директории |
| 2 | hisat2-build chr17.fasta chr17 | индексирует референсную последовательность, выдаёт несколько файлов .ht2 формата |
| 3 | hisat2 -x chr17 -U chr17clean.fastq --no-spliced-alignment --no-softclip > align.sam | строит выравнивание прочтений и референса (результаты сохранены в файл align.sam) |
Анализ выравниваний.
| № команды | Команда | Результат выполнения команды | |||
| 1 | samtools view align.sam -bo align.bam | переводит выравнивание с референсом в бинарный формат | |||
| 2 | samtools sort align.bam -T sort.txt -o sorted.bam | сортирует выравнивание чтений с референсом по координате в референсе начала чтения | |||
| 3 | samtools index sorted.bam | индексирует отсортированный .bam файл |
| № команды | Команда | Результат выполнения команды |
| 1 | samtools mpileup -uf chr17.fasta sorted.bam -o snp.bcf | cоздаёт файл в полиморфизмами в формате .bcf |
| 2 | bcftools call -cv snp.bcf -o snp.vcf | создаёт файл со списком отличий между референсом и прочтениями в формате .vcf - файл snp.vcf |
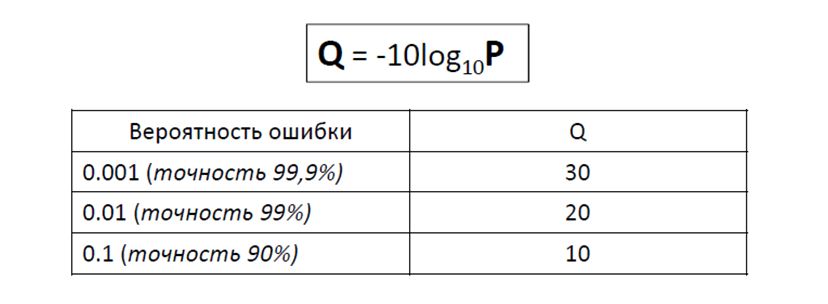
По результатам выдачи команды bcftools было найдено 58 полиморфизмов, из которых 4 инделя и 54 snp. Глубина покрытия и качество чтений сильно варьируются, но примерно треть полиморфизмов имеет покрытие выше 100 и качество выше 200, что является хорошим показателем.
| Позиция | Тип полиморфизма | Референс | Прочтение | Глубина покрытия | Качество чтений |
| 44788310 | замена | G | A | 74 | 221.999 |
| 79534241 | делеция | ATCTTTCT | ATCT | 4 | 13.657 |
| 44554051 | делеция | aaataataataataataataat | aaataataataataataat | 1 | 22.4955 |
Аннотация SNP.
Задача: с помощью программы annovar проаннотировать полученные snp, используя
базы данных: refgene, dbsnp, 1000 genomes, GWAS, Clinvar.
Команда: perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4
/nfs/srv/databases/ngs/s.kozyulina/snp.vcf > /nfs/srv/databases/ngs/s.kozyulina/snp.avinput
Выдача: файл, необходимый для работы annovar.
Поиск по базам данных:
intronic - 49
exonic - 3
intergenic - 1
UTR3 - 5
SNP также разделяются на het и hom замены: 20 het и 38 hom. Можно заметить, что подавляющее большинство
замен приходится на интроны. Это можно объяснить тем, что замены в интронах не подвергаются отбору и сохраняются,
так как не влияют на конечный продукт гена.
| Координата | Ген | Тип замены | было -> стало | Качество чтений | Глубина покрытия |
| 62007498 | CD79B | синонимичная | A -> G | 221.999 | 63 |
| 79589242 | NPLOC4 | синонимичная | G -> A | 21.999 | 103 |
| 79596811 | NPLOC4 | синонимичная | C -> T | 221.999 | 48 |
Таблица со всеми snp и их характеристиками, полученными выше: results.xlsx