Отчет по пр. 13
Сравнение выравнивания одних и тех же последовательностей тремя разными программами
Выравниваемые последовательности(AC):
A0A013WNS5; A0A031G1M3; A0A031HMD5; A0A031HPD9; A0A066WIK4
Программы для выравнивания:
muscle; mafft; probcons
Пояснение:
одинаковые участки - идентичные участки выравниваний (идентичные не только по а.к-те, но и по номерам ам.к-лот в последовательностях)
одинаковые блоки - блоки лежащие в одинаковых участках, то есть идентичные участки идентичных участков выравниваний (идентичные не только по а.к-те, но и по номерам ам.к-лот в последовательностях), начинающиеся функционально одинаковой а.к-той (вес в матрице замен > 0) для всех последовательностей выравнивания, оканчивающиеся так же, а выровненные последовательности внутри такого участка имеют высокое сходство (большое количество столбцов выравнивания с функционально одинаковой а.к-той, и отсутствие больших промежутков выравнивания без таких столбцов) и не содержат гэпов
Результаты:
muscle/mafft:
одинаковые участки: (42-414)=(48-420); (421-441)=(427-447); (449-490)=(457-498); (495-500)=(503-508); (507-515)=(516-524)…
одинаковые блоки: (44-83)=(50-89); (90-111)=(96-117); (117-146)=(123-152); (160-343)=(166-349); (353-414)=(359-420)…
одинаковые колонки вне блоков - все что попадает в «одинаковые участки» и не попадает в «одинаковые блоки»: (42-43)=(48-49); (84-89)=(90-95) и тд
muscle/probcons:
одинаковые участки: (40-111)=(40-111); (114-148)=(114-148); (159-414)=(159-414); (418-442)=(418-442); (448-490)=(448-490)…
одинаковые блоки: (44-83)=(44-83); (90-111)=(90-111); (117-146)=(117-146); (160-343)=(160-343); (353-414)=(353-414)…
одинаковые колонки вне блоков - все что попадает в «одинаковые участки» и не попадает в «одинаковые блоки»: (40-43)=(40-43); (84-89)=(84-89) и тд
Обсуждение:
Сравнение сделано вручную с помощью VerAlign. На участках 1-490 процент совпадения у muscle и mafft = 88.3%; у muscle и probcons = 86.9%. Отличие незначительное и в другой раз стоит взять выборку последовательностей больше, и делать сравнение программным методом. В целом результат выравнивания более менее похожий
Сравнение выравнивания одних и тех же последовательностей: совмещение пространственных структур и программой MSA (muscle)
Выравниваемые последовательности (pdb id): 1ar4; 1d5n; 1gn3 (для всех только цепь А)
одинаковые участки: (3-48)=(3-48); (80-98)=(72-90); (107-162)=(99-154); (178-220)=(169-211)
одинаковые блоки: (3-48)=(3-48); (80-97)=(72-89); (110-144)=(102-136); (150-160)=(142-152); (178-218)=(169-209)
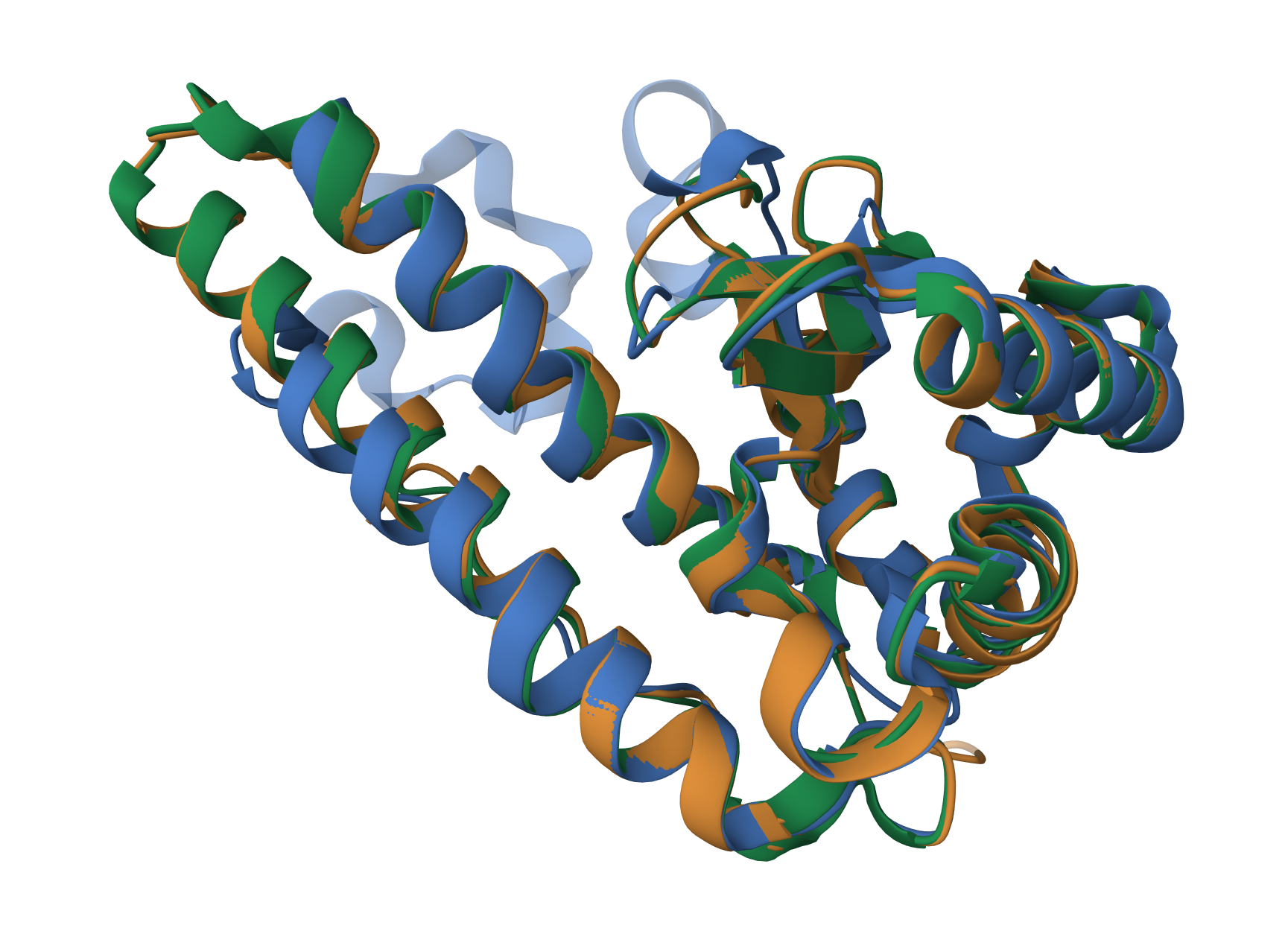
Совмещение пространственных структур
Обсуждение:
Сначала про совмещение структур: лучше всего совмещается на a-спиралях и b-листах, тк там много остова, который имеет +- одинаковую структуру. Визуально совмещение не выглядит очень плохо. За референсный брался 1ar4, и процент идентичности = 45% и 62% для 1d5n и 1gn3 соответственно.
Выравнивания через muscle и совмещение пространственных структур совпадают на 71.7% на всей длине выравнивания (223). Это немного ниже чем в предыдущем пункте, возможно из-за сильных отличий методик. Но вообще, это странно - делать выводы по такому количеству данных
Описание Muscle5
Этот програмный метод множественного выравнивания белковых и нуклеотидных последовательностей был представлен в марте 2004 года Робертом Эдгаром.
Алгоритм состоит из 3-х этапов:
1) Для каждой пары последовательностей находятся n-меры и по ним строится матрица расстояний, по которой далее строится направляющее древо. Далее от листьев идет выравнивание готовых пар между собой, потом пар пар, пар пар пар, пар пар пар пар, пар пар пар пар паахахха. И так далее до области рядом с корнем дерева.
2) Строится более точное дерево, где учитывается неравновероятность замены различных нуклеотидов/аминокислот. Снова используется прогрессивное выравнивание, но тк некоторые поддеревья не изменились, все проходит быстрее.
3) Второе дерево разрезается на два поддерева, и каждое выравнивается отдельно. Далее поддеревья выравнивают друг с другом, и если результат получается лучше, чем был, то работаем с этим выравниванием. Этап повторяется нужное количество раз, причем дерево режется все ближе к корню. Лучший полученый результат и выдается программой.
Отличительный этап третий. Он позволяет сильно уточнить результат, снизив ошибки на этапе попарных выравниваний входных последовательностей
На этом все