Филогенетическое дерево и его реконструкция

- Реконструкция дерева вручную по скобочной формуле. Скобочная формула, задающая гипотетическую модель эволюции гена, имеет следующий вид:
- Описание ветвей дерева через разбиения множества листьев.
- Получение мутантных последовательностей, соответствующих листьям дерева из упражнения 1.
- Реконструкция филогенетического дерева по набору последовательностей тремя разными методами.
- Бутстреп-анализ выравнивания мутированных последовательностей A,B,C,D,E и F. В отчете по этому упражнению опишите, совпало ли консенсусное дерево по топологии с реальным деревом, а также каковы бутстреп-значения внутренних ветвей консенсусного и реального деревьев. Проиллюстрируйте свои выводы выдержками из выходного файла.
- Визуализация скобочных формул с помощью fdrawtree.
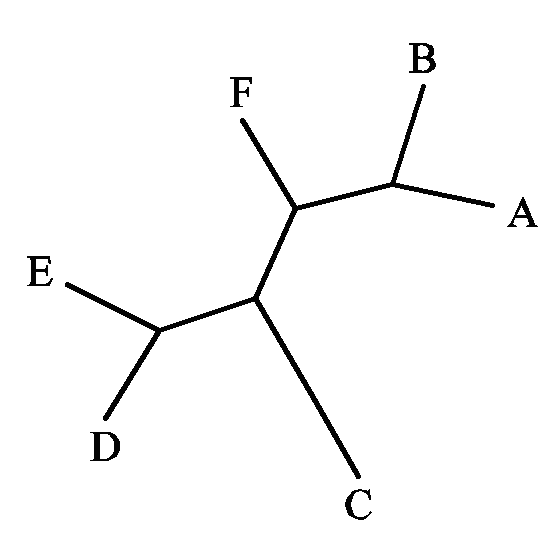
Используя эту формулу, вручную в графическом редакторе Paint создали изображение соответствующего ей филогенетического дерева. Заметим, что это укорененное дерево. Красным отмечены внутренние ветви (см.следующее упражнение)

|
Расстояние до разных листьев может быть разной длины (обратим внимание на кладу CDE), поскольку в качестве эволюционного
расстояния, отраженного длиной ветвей,
в нашей модели фигурирует число мутаций (замен), а не время эволюции.
В любом бинарном неукорененном дереве, содержащем N листьев, всегда имеется 2N–3 ветви. Следовательно, в дереве из шести листьев будет 9 ветвей, из них шесть внешних ветвей и три внутренних. Внешние ветви соединяют какой-либо лист с остальными элементами дерева; такие ветви описываются тривиально, и они есть в любом дереве из шести листьев, поэтому соответствующие разбиения здесь не приводятся. Каждая внутренняя ветвь разбивает множество всех листьев дерева, в нашем случае множество ABCDEF, на два подмножества - листьев, расположенных с одного и с другого конца ветви. Ниже приведено описание ветвей дерева из упражнения 1 в виде таблицы разбиений множества листьев каждой ветвью. Точками обозначены ветви, принадлежащие одному подмножеству, звездочками - другому (не важно, какому). Заметим, что наше дерево укорененное, поэтому в нем появляется еще один узел - корень. а также еще одна внутренняя ветвь (см.красные ветви на иллюстрации к упражнению 1)
Листья A B C D E F Ветви 1 . . * * * * 2 . . * * * . 3 . . . * * . 4 . . . . . *
Применим модель эволюции, описанную скобочной формулой в упражнении 1, к прокариотическому гену aspC. Для этого с помощью программы msbar создадим мутантные последовательности, соответствующие всем узлам и листьям дерева в том порядке, какой диктует нам направление эволюции. К примеру, чтобы "создать" мутанта A, сначала получим через msbar последовательность некоего общего для A и B предка, а потом только из него получим A, а не внесем сразу все мутации в последовательность aspC. В нашей скобочной формуле расстояния, отделяющее одну последовательность от другой, даны в количестве замен на одну сотню нуклеотидов. Для работы с msbar требуется задать абсолютное число замен в последовательности. Ген aspC имеет длину 1331 нуклеотид. Поэтому приведенные в скобочной формуле числа умножим на 1331 и разделим на 100. Полученные результаты округлим до целого (число замен не может быть дробным). Абсолютные количества замен используем при написании скрипта, в результате работы которого получается файл со всеми мутантными последовательностями (только A,B,C,D,E,F, но не последовательности узлов и не исходный ген!). Текст скрипта можно посмотреть, щелкнув сюда. Добавим, что в полученном файле ABCDEF мы вручную изменили названия последовательностей на необходимые (A-F).
Полученный в предыдущем упражнении файл ABCDEF.fasta подадим на вход трем программам, реализующим различные алгоритмы реконструкции филогенетических деревьев. Для создания дерева по набору последовательностей методом максимального правдоподобия (AML - Analysis by Maximum Likelihood) используем программу fdnaml. Для ее работы не требуется предварительныого вычисления матрицы попарных расстояний; программа реконструирует неукорененное дерево следующего вида:
+--------------B | | +----------------------------------F | | 1--------------4 +-------E | | +------3 | +-------------2 +---------D | | | +---------C | +--------A
Другими двумя алгоритмами реконструкции деревьев мы уже пользовались во втором семестре (см.отчет по практикуму "Филогенетические деревья, реконструированные разными способами". Это алгоритмы UPGMA и Neighbor-joining; первый строит укорененные деревья, второй - неукорененные. Оба алгоритма алгоритмов работают только с матрицей попарных расстояний, поэтому их использование требует сначала подсчета попарных расстояний между последовательностями с помощью программы fdnadist.
Linux-реализацией пакета Phylip мы пользуемся впервые; оба алгоритма "зашиты" в одну программу fneighbor, вид алгоритма задается значением параметра treetype (UPGMA - u; Neighbor-joining - по умолчанию - n). Полученные деревья имеют следующий вид:
+--------------B ! ! +-----------------------------------F 1--------------2 ! ! +--------C ! +---------------3 ! ! +---------D ! +-------4 ! +--------E ! +-------A |
| +-----------A +----------------2 ! +-----------B +---4 ! ! +------------C ! +--------------3 --5 ! +--------D ! +----1 ! +--------E ! +-------------------------------F |
Использование параметра ttratio При получении деревьев с помощью трех перечисленных выше алгоритмов мы не меняли никаких параметров программ fdnaml и fdnadist, кроме параметра ttratio. Данная опция, судя по справкам к названным программам, означает отношение числа транзиций к числу трансверсий в последовательностях, поданных на вход. По умолчанию значение ttratio равно двум, так как в природе мутации по типу транзиций встречаются чаще (транзиция - смена основания на однотипное - пурин на пурин, пиримидин на пиримидин, такая замена слабее меняет геометрию спирали ДНК, чем трансверсия - замена на основание другого типа; по этой причине трансверсии реже обнаруживаются репарационными системами клетки и реже элиминируются). Однако происхождение наших мутантых последовательностей, соответствующих листьям дерева из упражнения 1, - искусственное. Они были созданы из последовательности гена aspC с помощью программы msbar. Последняя делает замены в последовательности случайным образом, не отличая транзиции от трансверсии, поэтому в мутантах число тех и других замен примерно одинаково. По данной причине мы изменили значение параметра ttratio, сделав его равным 1; такое значение больше соответствует действительности, что обещает меньше ошибок при реконструкции дерева.
Анализ деревьев, полученных с помощью различных методов.
При выполнении предыдущих заданий мы получили четыре дерева: одно нарисовали "вручную" по скобочной формуле, еще три получили с помощью различных алгоритмов реконструкции по набору мутатных последовательностей, созданных согласно той же скобочной формуле. Сначала сравним эти деревья между собой, особенно уделяя внимание тому, насколько реконструированные деревья соответствуют первому - "правильному" с точки зрения заданной скобочной формулы. Для этого воспользуемся стандартной процедурой сравнения ветвей деревьев как разбиений множеств их листьев (см. упражнение 2).
A B C D E F Дерево "вручную" Дерево методом AML Дерево Neighbor-Joining Дерево UPGMA Тип дерева укорененное неукорененное неукорененное укорененное Ветви 1 . . * * * * + + + + 2 . . * * * . + + + + 3 . . . * * . + + + + 4 . . . . . * + - - + |
однако неукорененные деревья (AML и NJ) отображают ветви клады AB как ветви разной длины. Такие погрешности можно связать со
случайными событиями при "мутировании" с помощью msbar. Обе последовательности - A и B - создавались из одной (AB), при этом было
произведено одинаковое число замен. Однако число однотипных замен (например, G на G) и замен в том же сайте (G на C, а потом опять
на G или другой нуклеотид) могло быть разным, что отразилось на эволюционных расстояниях, визуализируемых длиной ветвей дерева.
С Online-версией пакета Phylip мы знакомились во втором семестре; в том числе с программами-визуализаторами drawtree и drawgram. Сейчас, в порядке ознакомления с Linux-версией того же пакета создадим изображение полученного в прошлом упражнении дерева с помощью fdrawtree. Эта программа визуализирует данную на вход скобочную формулу в виде неукорененного дерева в Postscript-формате. В результате получаем такое изображение:
|
Для интереса воспользуемся и Linux-аналогом программы drawgram - fdrawgram. Эта программа строит укорененные деревья. Вот что мы получили, подав ей на вход скобочную формулу из файла consensus.treefile (см. прошлое упражнение).

|
©Ганчарова Ольга