Комплексы ДНК-белок
 Предсказание вторичной структуры заданной тРНК.
Предсказание вторичной структуры заданной тРНК.
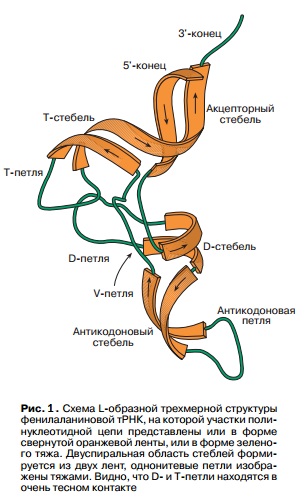
В первом задании необходимо было предсказать вторичную структуру тРНК с
помощью различных программ. На рис.1 представлена структура тРНК, а
также принятые названия для различных участков цепи. Мы будем
использовать две программы: einverted и RNAfold.
Программа
einverted из пакета EMBOSS позволяет найти инвертированные
участки в нуклеотидных последовательностях. Программа получает на вход
последовательность нуклеотидов (USA), в моем случае 2dxi.fasta. На выход
получаем два файла:
sequence.fasta и
sequence.inv. В первом записаны
найденные комплементарные участки, а во втором найденные водородные
связи в пределах этих участков. Я попыталась найти с ее помощью
возможные комплементарные участки в последовательности исследуемой тРНК
(PDB ID:2dxi). Так как при стандартных параметрах программа ничего не
находила, были попытки подобрать специальные параметры для получения
предсказания, наиболее близкого к реальной структуре. Такими параметрами
оказались:
gap penalty[12]: 9;
minimum score threshold[50]: 20;
match score[3]: 4;
mismatch score[-4]:-5.
Несмотря на попытку подогнать результаты, даже в этом случае они были
ужасными по сравнению с программой
find_pair. Программа einverted нашла только 4 пары, и те неверные, хотя и
наиболее близкие к реальности: все найденные связи сдвинуты на один нуклеотид.
Также хочется отметить, что einverted почему-то заменила все уридины на тимины
в файлах sequence.inv и sequence.fasta. Помимо всех этих минусов, программа также
неспособна обнаруживать неканонические взаимодействия, что заведомо вносит ошибки
в результат.
Следующей использовалась программа
RNAfold из пакета Viena Rna Package,
которая реализует алгоритм Зукера. Была использована следующая команда:
cat my.fasta | RNAfold --MEA > rna_fold.fasta
Таким образом был получен файл
rna_fold.fasta, в
котором первую строчку представляет собой последовательность, а следующие строчки
— предсказанная структура MFE. Квадратными совпадающими скобками
обозначена связь пары оснований, и далее
указана свободная энергия в ккал/моль. Точка в обозначает нуклеотид, не образующий
пару. Кроме того с помощью той же программы были получены файлы 2dxi_ss.ps
и 2dxi_dp.ps, которые затем были конвертированы в формат jpeg и представлены
на рис. 1 и 2 соответственно. Сравнение результатов с выдачей программы
find_pair представлены в табл. 1.
Таблица 1. Реальная и предсказанная вторичная структура
тРНК из файла 2dxi.pdb
| Участок структуры |
Позиции в структуре (по результатам find_pair) |
Результаты предсказания с помощью einverted |
Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель |
5' ----------------> 3'
C:501[G]---[C]572:C
C:502[G]---[U]571:C
C:503[C]---[G]570:C
C:504[C]---[G]569:C
C:505[C]---[G]568:C
C:506[C]---[G]567:C
C:507[A]---[U]566:C
Всего 7 пар
|
5' ----------------> 3'
(лучший неверный результат)
C:503[C]---[G]569:C
C:504[C]---[G]568:C
C:505[C]---[G]567:C
C:506[C]---[G]566:C
C:507[A]---[U]565:C
Всего 0 верных пар
|
5' ----------------> 3'
C:501[G]---[C]572:C
C:502[G]---[U]571:C
C:503[C]---[G]570:C
C:504[C]---[G]569:C
C:505[C]---[G]568:C
C:506[C]---[G]567:C
C:507[A]---[U]566:C
Всего 7 пар
|
| D-стебель |
C:510[G]---[C]525:C
C:511[U]---[A]524:C
C:512[C]---[G]523:C
C:513[U]---[G]522:C
Всего 4 пары
|
Всего 0 пар
|
C:510[G]---[C]525:C
C:511[U]---[A]524:C
C:512[C]---[G]523:C
C:513[U]---[G]522:C
Всего 4 верных пары
C:509[C]---[G]526:C
+1 лишняя пара
|
| T-стебель |
C:549[G]---[C]565:C
C:550[G]---[C]564:C
C:551[G]---[C]563:C
C:552[G]---[C]562:C
C:553[G]---[C]561:C
Всего 5 пар
|
Всего 0 пар
|
C:549[G]---[C]565:C
C:550[G]---[C]564:C
C:551[G]---[C]563:C
C:552[G]---[C]562:C
C:553[G]---[C]561:C
Всего 5 пар
|
| Антикодоновый стебель |
C:538[A]---[C]532:C
C:539[G]---[C]531:C
C:540[G]---[C]530:C
C:541[C]---[G]529:C
C:542[C]---[G]528:C
C:543[G]---[C]527:C
C:544[A]---[G]526:C
Всего 7 пар
|
Всего 0 пар
|
C:539[G]---[C]531:C
C:540[G]---[C]530:C
C:541[C]---[G]529:C
C:542[C]---[G]528:C
C:543[G]---[C]527:C
Всего 5 пар
|
| Общее число верно найденных пар нуклеотидов |
23 пары
|
0 пар
|
21 пара
|
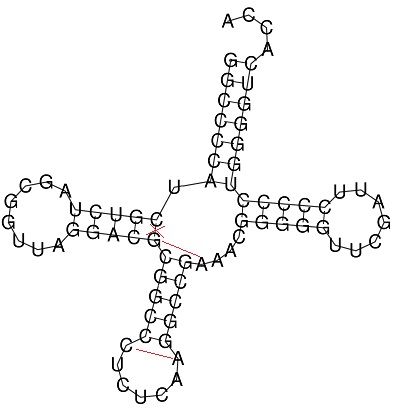 Рис. 1. Предсказанная RNAfold структура тРНК. Рис. 1. Предсказанная RNAfold структура тРНК.
Крестиком
обозначена неправильно
найденная связь; линиями —
ненайденные, но существующие связи |
 Рис. 2. Полученная матрица Рис. 2. Полученная матрица
|
В общем, хочется сказать, что из всех программ лучше всего находит водородные связи между нуклеотидами
find_pair, а хуже
всех —
einverted. Программа
RNAfold тоже довольно хорошо справляется с поставленной задачей, но
все-таки результаты ее работы нужно проверять. Зато можно сразу получить примерную структуру тРНК в плоскости, что, конечно, помогает
легче ее анализировать.
Поиск ДНК-белковых контактов в заданной структуре. В этом задании необходимо
было изучить ДНК-белковые контакты в заданной структуре с PDB ID: 1by4. Для начала
с помощью команды
define были определены три множества атомов: множество атомов
кислорода 2'-дезоксирибозы (set1), множество атомов кислорода в остатке фосфорной
кислоты (set2) и множество атомов азота в азотистых основаниях (set3). Затем был
создан скрипт (
текст скрипта) с определениями этих множеств.
Нажав на выдру слева, вы увидите исходный комплекс. Затем используйте кнопку
"Start script 1" (1 раз!) и "Resume".
Изучим теперь различные контакты между белком и ДНК. Договоримся, что:
1.полярные атомы — атомы кислорода и азота;
2.неполярные атомы — атомы углерода, фосфора и серы;
3.полярный контакт — ситуация, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5Å;
4.неполярный контакт — пара неполярных атомов на расстоянии меньше 4.5Å.
С учетом этих договоренностей, с помощью результатов предыдущего практикума (по определению атомов
большой и малой бороздок) и с помощью уже определенных множеств атомов (sets) были
найдены различные контакты между ДНК и белком (
текст скрипта). Число разных контактов приведено
в таблице 2. Для просмотра полученных структур в апплете используйте кнопку
"Start script 2" (1 раз!) и затем "Resume".
Таблица 2. Контакты разного типа в комплексе 1by4.pdb
| Контакты атомов белка с |
Полярные |
Неполярные |
Всего |
| остатками 2'-дезоксирибозы |
13 |
7 |
20 |
| остатками фосфорной кислоты |
51 |
45 |
96 |
| остатками азотистых оснований со стороны большой бороздки |
4 |
11 |
15 |
| остатками азотистых оснований со стороны малой бороздки |
1 |
7 |
8 |
Согласно полученным данным (см. таблицу 2) больше всего контактов образуется
с участием остатков фосфорной кислоты; на втором месте — остатки 2'-дезоксирибозы;
на третьем — остатки азотистых оснований со стороны большой бороздки.
Заметим, что контактов с участием остатков азотистых оснований со
стороны малой бороздки было найдено меньше всего. Таким образом, можно заключить, что остов ДНК больше
склонен к образованию связей, чем остатки азотистых оснований. Так же
вполне логичным кажется то, что остатки азотистых оснований со стороны большой бороздки
образуют большее количество связей, чем остатки азотистых оснований со стороны малой бороздки.
Думаю, это объясняется стерическим фактором (большей доступностью
атомов остатков азотистых оснований со стороны большой бороздки).
Использование программы nucplot для получения популярной схемы ДНК-белковых контактов.
Так как программа работает только со старым форматом pdb, сначала была использована
команда 1, а затем уже команда 2:
(1) remediator --old 1by4.pdb > 1by4_old.pdb
(2) nucplot 1by4_old.pdb
На выход
nucplot дает несколько файлов: *.hb2, *.nb2, hbplus.rc, *.bond, nucplot.ps, из которых
нас интересуют только последние два. Файл 1by4_old.bond содержит информацию о всех найденных
связях, а файл nucplot.ps — ту самую популярную схему ДНК-белковых контактов. Файл nucplot.ps
был конвертирован в JPEG постранично (рис. 3-6). Хочется отметить, что программа нашла не только
определенные нами полярные и неполярные контакты, но и различные другие (например, между углеродом
аминокислотного остатка белка и кислородом дезоксирибозы). Как я понимаю, в этом случае критерием
наличия какого-либо взаимодействия служило то, что те или иные атомы находятся на расстоянии < 3,5Å.
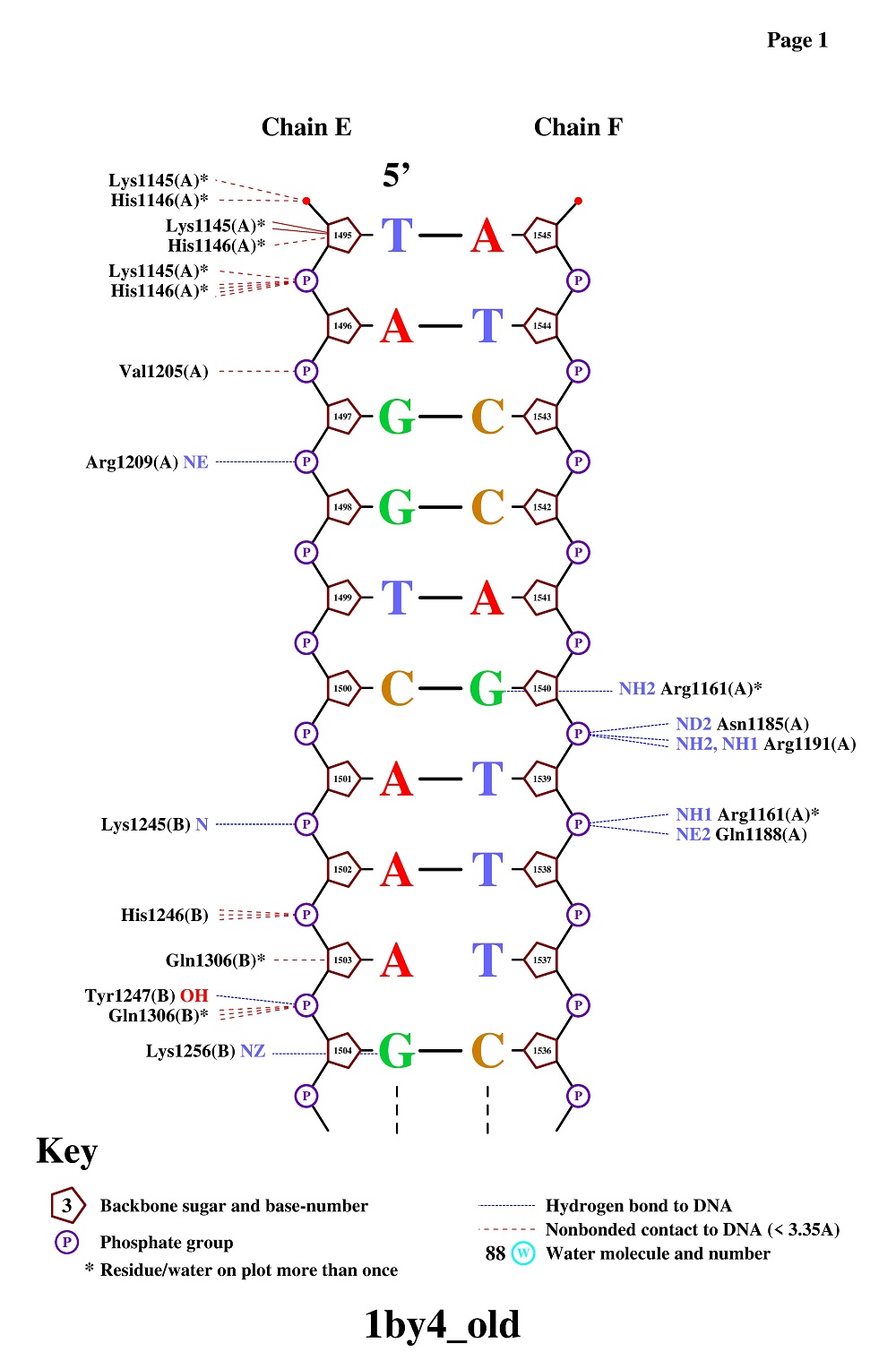 Рис. 3. Популярная схема ДНК-белковых контактов (стр.1) Рис. 3. Популярная схема ДНК-белковых контактов (стр.1)
|
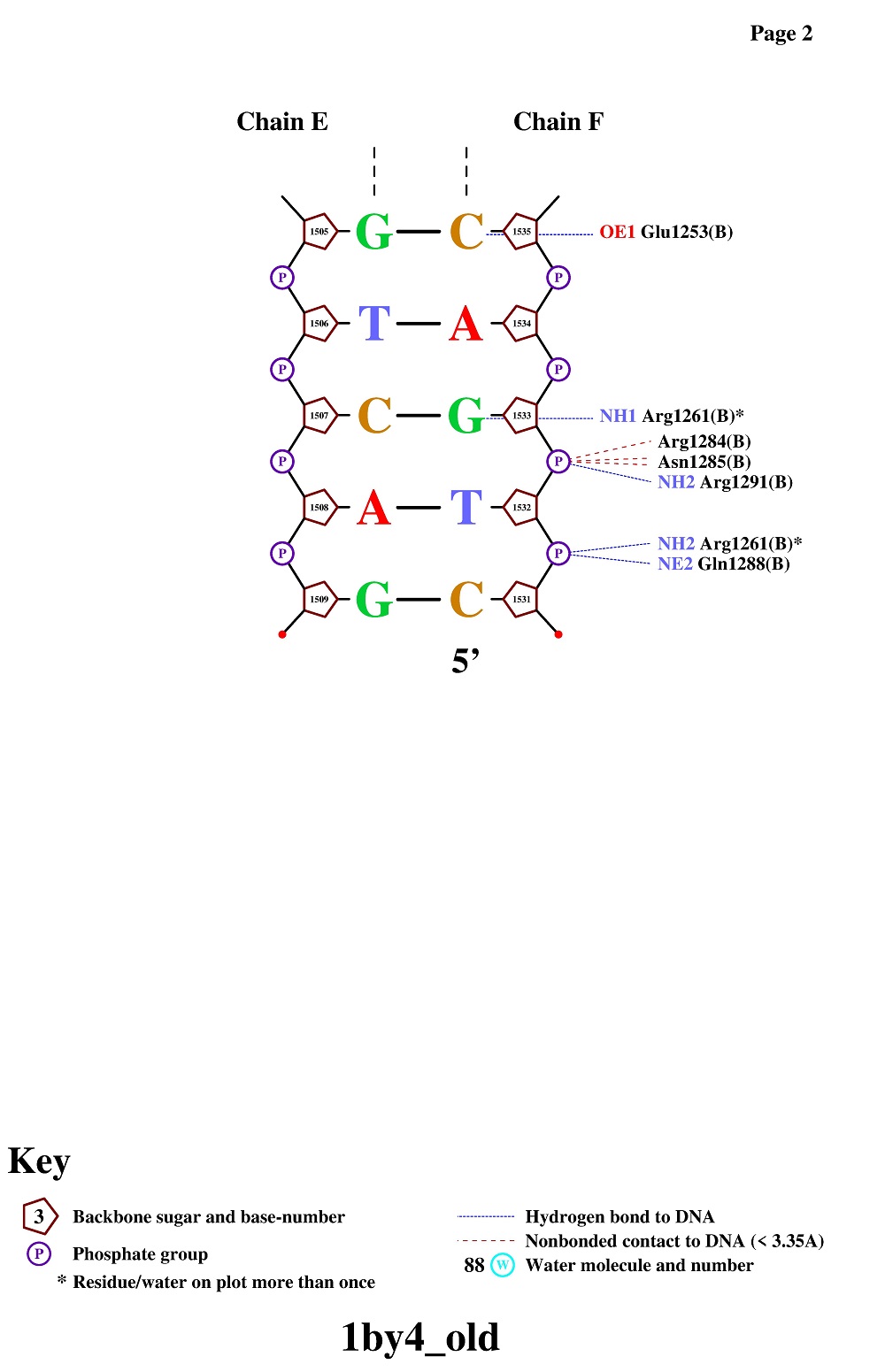 Рис. 4. Популярная схема ДНК-белковых контактов (стр.2) Рис. 4. Популярная схема ДНК-белковых контактов (стр.2)
|
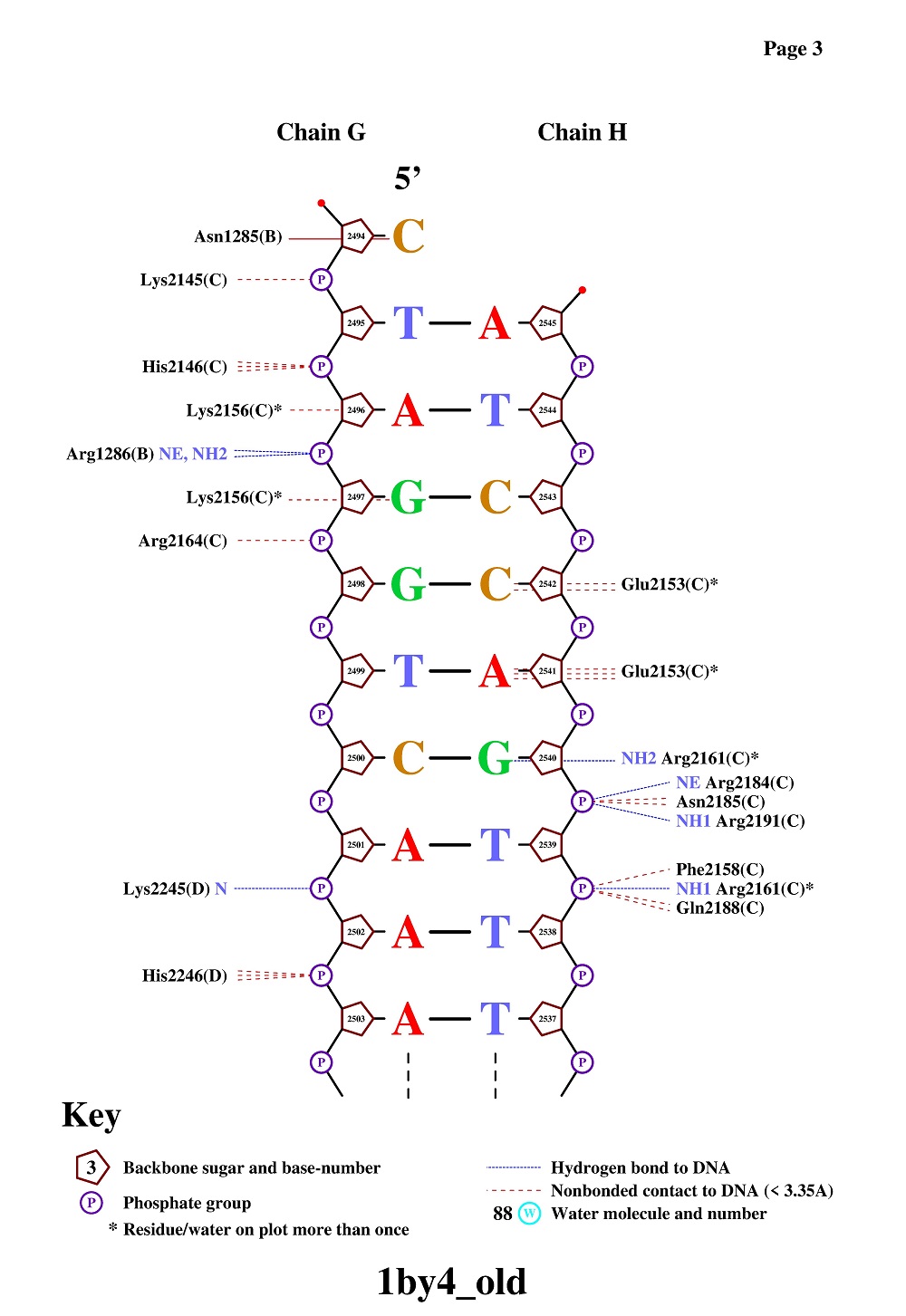 Рис. 5. Популярная схема ДНК-белковых контактов (стр.3) Рис. 5. Популярная схема ДНК-белковых контактов (стр.3)
|
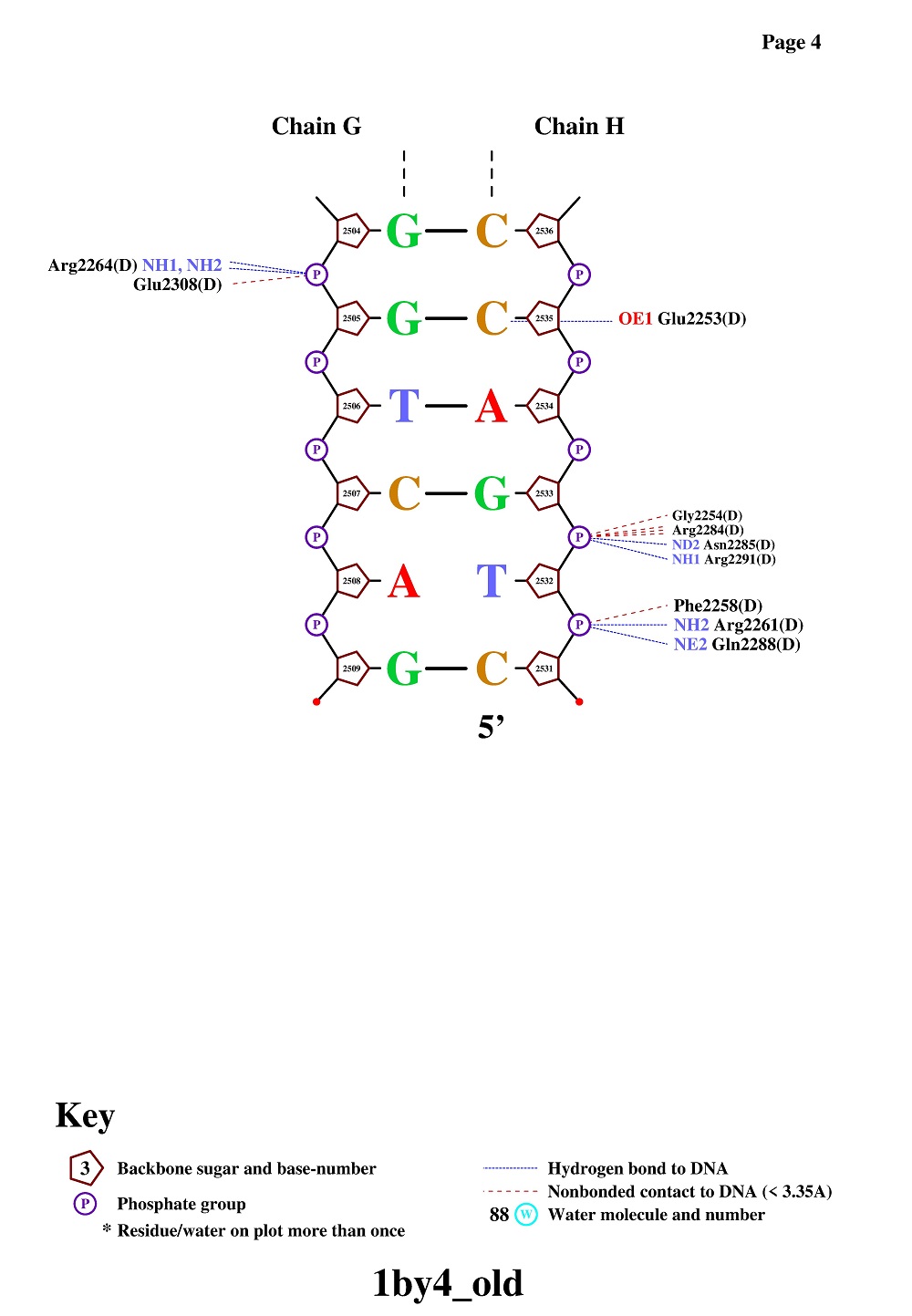 Рис. 6. Популярная схема ДНК-белковых контактов (стр.4) Рис. 6. Популярная схема ДНК-белковых контактов (стр.4)
|
Если учитывать не только наши "полярные" и "неполярные" контакты, а вообще все контакты, которые
нашла программа nucplot, то больше всего связей образовывает HIS1146 цепи A (а именно 7
связей). Так как этот аминокислотный остаток образует больше всего взаимодействий, его определение
наиболее важно для распознавания последовательности ДНК. На рис. 8 представлено изображение одной из
найденных полярных связей для этого остатка ([HIS]1146:A.O --- [DA]1496:E.OP1), полученное в Jmol.
Аминокислотным остатком, наиболее важным для распознавания последовательности ДНК, я бы назвала
[GLU]2153 цепи C, т.к. он (один из немногих) образует связь с двумя азотистыми основаниями, а не с
остовом, при этом в сумме этот остаток образует 5 связей (в то время как среднее число контактов
около 2-3). На рис. 7 приведено изображение одного из полярных контактов ([GLU]2153:C.CG --- [DA]2541:H.C8),
полученное в Jmol.
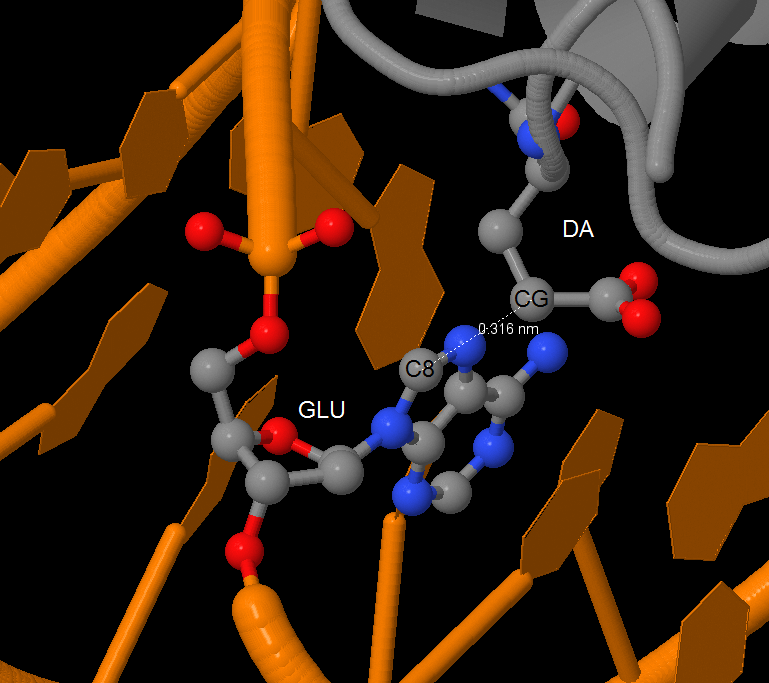 Рис. 7. Неполярный контакт Рис. 7. Неполярный контакт
[HIS]1146:A.O --- [DA]1496:E.OP1 и его окружение.
|
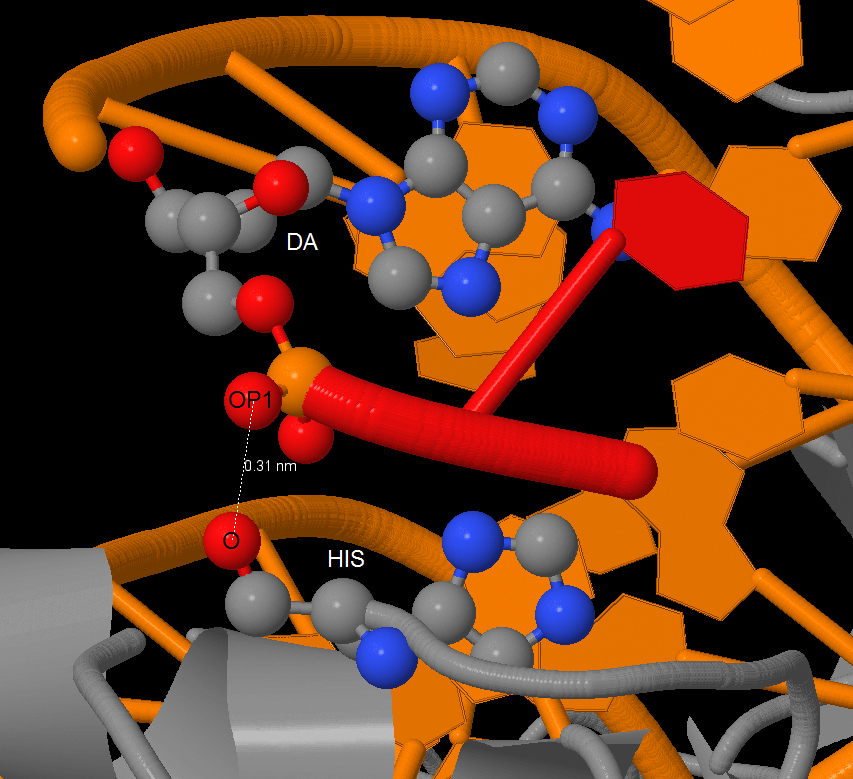 Рис. 8. Полярный контакт Рис. 8. Полярный контакт
[GLU]2153:C.CG --- [DA]2541:H.C8 и его окружение.
|