Остальные пункты первого задания
6. С помощью
seqret перевести выравнивание из fasta-формата в .msf.
На вход подается выравнивание:
alignment.fasta:
seqret alignment.fasta msf::alignment.msf
а на выходе получаем то же выравнивание, но уже в формате .msf:
alignment.msf.
7. С помощью команды
infoalign выдать в выходной поток число
совпадающих букв между второй последовательностью выравнивания и всеми остальными
(на выходе только имя последовательности и число). На вход подавался файл с
выравниванием пяти последовательностей:
aligns.msf:
infoalign aligns.msf info_align.txt -refseq 2 -only -name -idcount
Был получен
info_align.txt. Так как я не придумала,
как отменить запись в файл и направить выдачу в stdout, эта задача решалась в лоб:
cat info_align.txt
в результате чего получаем:
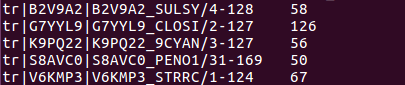 8.
8. C помощью команды
featcopy перевести аннотации особенностей в
записи формата .gb в табличный формат .gff. На вход подавалась уже упомянутая
ранее нуклеотидная последовательность
sequence.gb:
featcopy sequence.gb -auto
в результате чего был получен файл с названием по умолчанию:
sequence.gff, содержащий аннотации особенностей в формате
таблицы.
9. С помощью
extractfeat из одного файла с хромосомой в формате .gb
получить fasta файл с кодирующими последовательностями; (*) добавить в описание
каждой последовательности функцию белка (из поля product). На вход опять подавался
файл
sequence.gb:
extractfeat sequence.gb info.fasta -type CDS -describe product
На выходе получаем файл
info.fasta со всеми
кодирующими последовательностями из входного файла, а также с описаниями
функции каждого белка, например, (product="hypothetical protein").
10. С помощью
shuffleseq* перемешать буквы в данной нуклеотидной
последовательности. На вход подавался файл
gene.fasta
с последовательностью некоторого гена:
shuffleseq gene.fasta shuffled.fasta
и получаем на выходе файл:
shuffled.fasta.
* мною использовалась команда
shuffleseq, так как она входит в
EMBOSS, который мы изучаем, в отличие от
shuffle, которая входит в
biosquid.
Далее предлагалось проверить найдет ли blastn достоверные (e-value до 0.1)
сходные последовательности в нуклеотидном банке данных. Для этого был осуществлен
поиск blastn с параметрами по умолчанию. На рис. 1 приведены результаты поиска.
Рис. 1. Результаты поиска blastn для перемешанной
последовательности
Как видно на рисунке, было найдено 10 последовательностей с e-value меньше 0.1,
однако query cover очень низкий. К тому же, я бы скорее использовала пороговое
значение e-value меньше 0.001. Для исходной последовательности, очевидно, было
найдено большое количество находок с очень низким e-value (данные не приведены),
что подтверждает тот факт, что этот параметр позволяет отсеивать недостоверные
находки.
11. С помощью
cusp найти частоты кодонов в данных кодирующих
последовательностях. На вход подавался файл
info.fasta:
cusp info.fasta kodons.cusp
и в итоге был получен файл
kodons.cusp, содержащий
статистику по всем кодонам в исходных CDS.
12. С помощью
compseq найти частоты динуклеотидов в данной
нуклеотидной последовательности и сравнить их с ожидаемыми. На вход подавался
файл
gene.fasta:
compseq gene.fasta gene.compseq -word 2 -calcfreq
В выходном файле
gene.compseq содержатся частоты
динуклеотидов и их отношение к соответствующим ожидаемым частотам.
13. С помощью
tranalign выровнять кодирущие последовательности
соответственно выравниванию белков (их продуктов). На вход подавался файл
info.fasta:
слишком сложно, до свидания!

