Сайты связывания транскрипционного фактора в участке хромосомы человека
1. Контроль качества ридов
Для выполнения практикума мне был дан файл с ридами
chipseq_chunk41.fastq. Сначала я произвела оценку
ридов, чтобы понять, нужна ли очистка файла. Для этого
использовалась программа
FastQC:
$ fastqc chipseq_chunk41.fastq
Так был получен файл
.html
, содержащий информацию о ридах. Несмотря на то, что
низкокачественных ридов найдено не было ("Sequences flagged as poor
quality: 0"), усы мне показались слишком большими, поэтому я провела
очистку ридом программой
Trimmomatic , убрав риды с качеством ниже 50 и минимальной
длиной 36 п.н.:
$ java -jar /usr/share/java/trimmomatic.jar SE -phred33 chipseq_chunk41.fastq chipseq_chunk41_out.fastq TRAILING:50 MINLEN:36
В итоге был получен
.html файл с улучшенными ридами. В таблице 1 представлена
информация о ридах до и после чистки.
Таблица 1. Риды до и после чистки
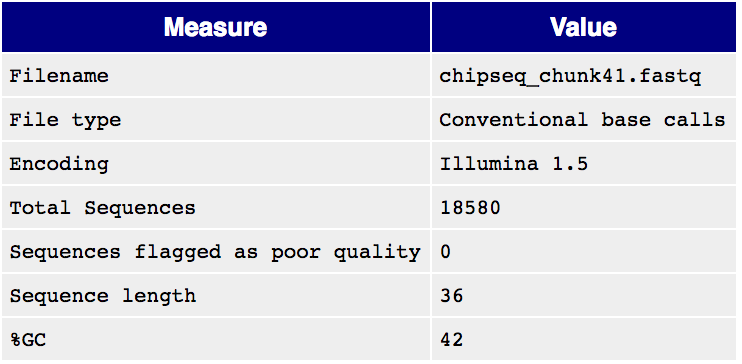 Рис. 1. Общая сводка по ридам до
чистки
Рис. 1. Общая сводка по ридам до
чистки |
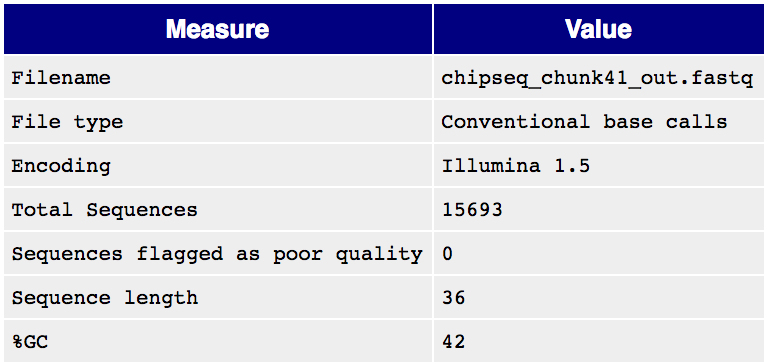 Рис. 2. Общая сводка по ридам после
чистки
Рис. 2. Общая сводка по ридам после
чистки |
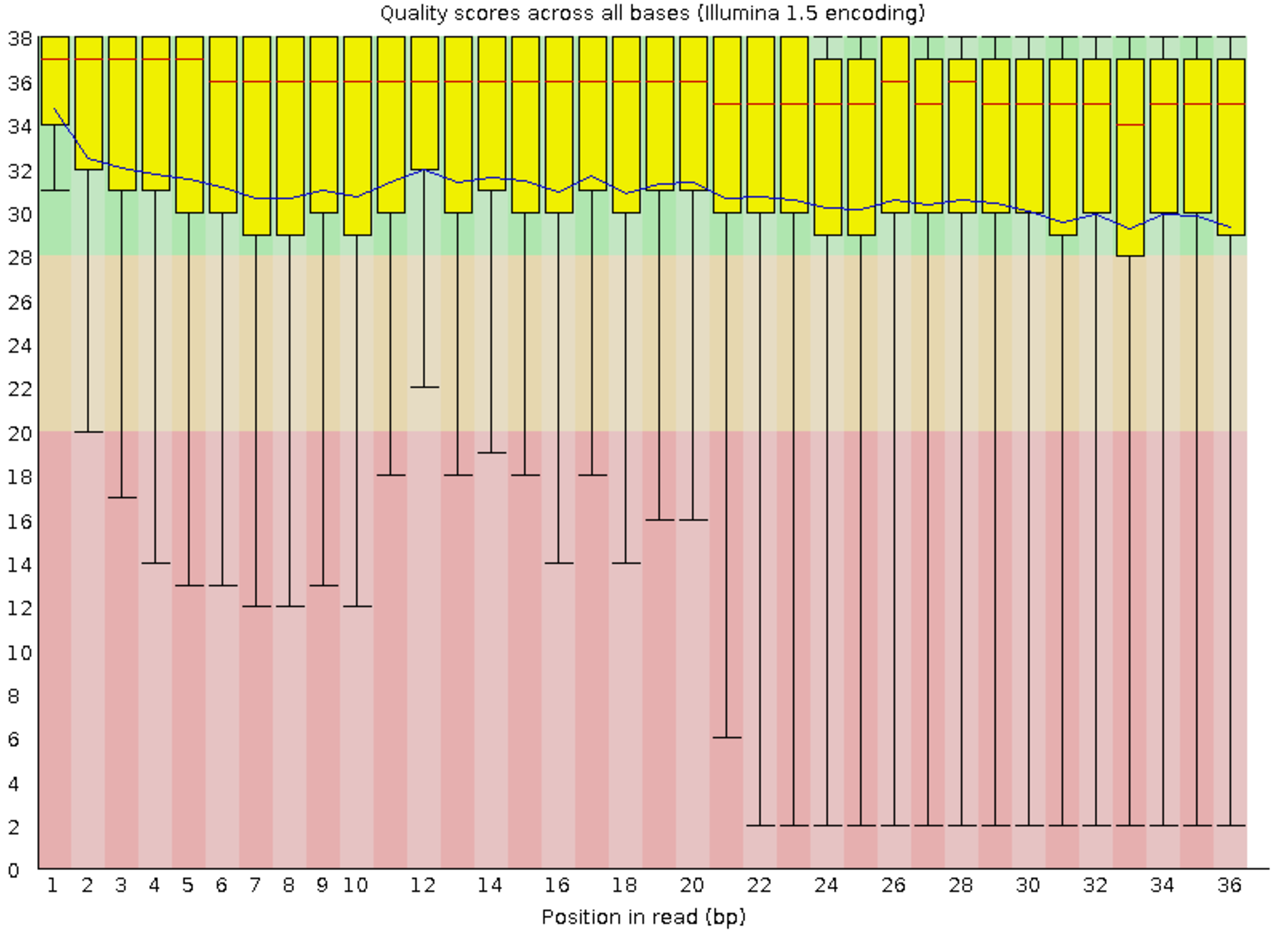 Рис. 3. Качество ридов до чистки
Рис. 3. Качество ридов до чистки |
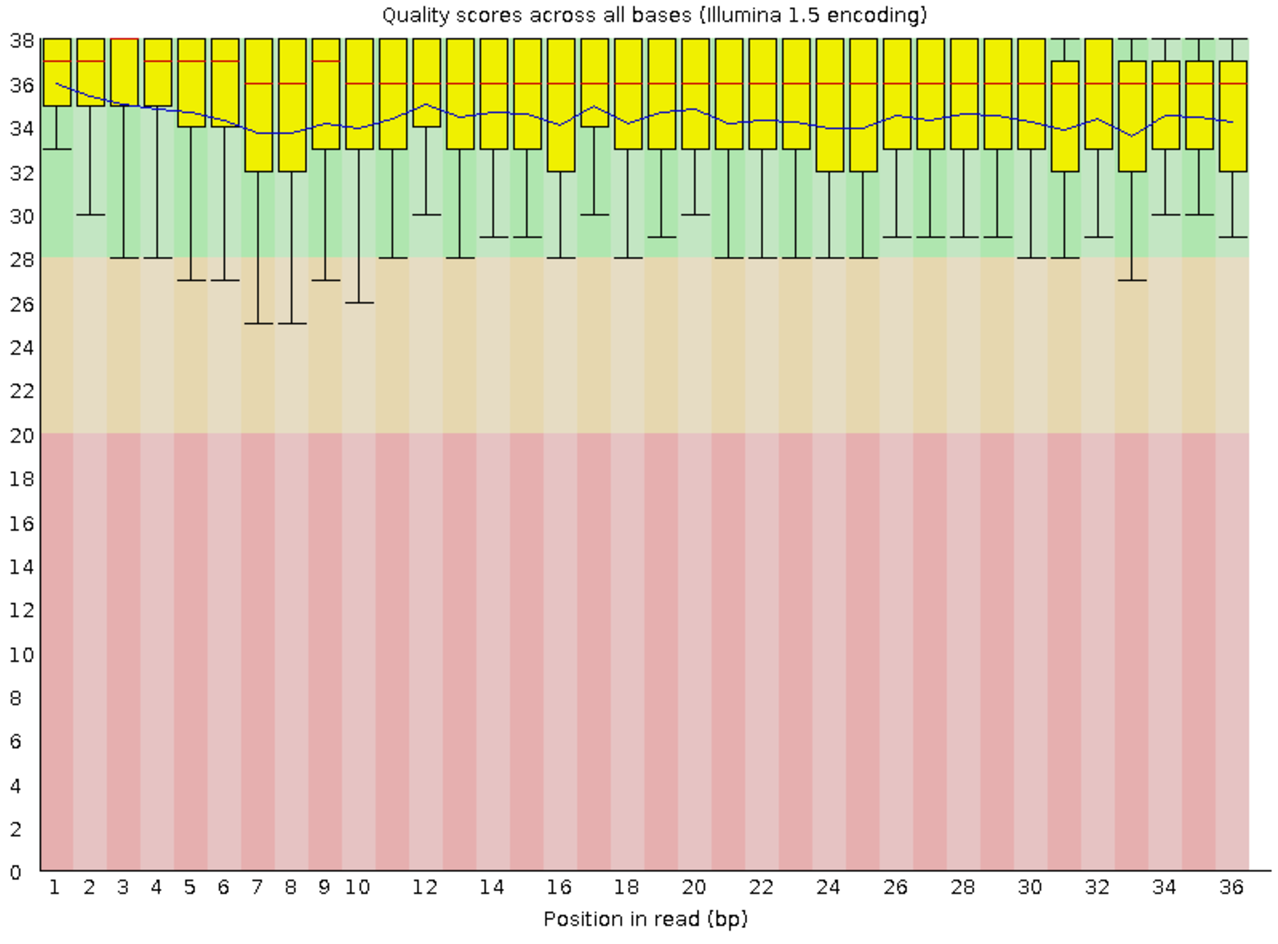 Рис. 4. Качество ридов после чистки
Рис. 4. Качество ридов после чистки |
Таким образом было удалено 15.54% ридов, качество оставшихся значительно
улучшилось. С полученным файлом производилась дальнейшая работа.
2. Картирование ридов на геном человека
Картирование выполнялось с помощью команды
BWA для
проиндексированного референса:
$ /srv/databases/ngs/seferbekova$ bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk41_out.fastq >
chipseq_chunk41.sam
Далее для анализа картирования было выполнено несколько команд. Сначала
с помощью пакета
samtools файл с выравниванием был переведен в
бинарный формат
.bam:
$ samtools view chipseq_chunk41.sam -bSo chipseq_chunk41.bam
Получившийся файл
chipseq_chunk41.bam был подан на вход другой
команде, чтобы отсортировать выравнивание ридов с референсом по
координате в референсе начала рида (параметр -T позволяет записать
временные файлы в файл chip_temp, т.к. иначе они выводятся в stdout):
$ samtools sort chipseq_chunk41.bam -T chip_temp -o chipseq_chunk41_sorted.bam
Далее полученный файл был проиндексирован:
$ samtools index chipseq_chunk41_sorted.bam
А затем я выяснила, сколько чтений откартировалось на геном:
$ samtools idxstats chipseq_chunk41_sorted.bam > chip.out
получив результат, записанный в файл
chip.out.
Таким образом, из 15693 ридов 14323 (91.27%) были откартированы на 22
хромосому, так что предполагаю, что риды с именно этой хромосомы были
предложены мне для анализа. При этом откартировались все 15693 рида.
3. Поиск пиков (peak calling)
Peak calling — компьютерный метод поиска областей генома,
на которые откартировалось большое число ридов из данных эксперимента
ChiP-Seq. В случае ТФ эти области соответствуют сайтам связывания ТФ.
Для поиска пиков использовалась программа
MACS
(Model-based Analysis of Chip-Seq) — одна из самых популярных
программ для анализа данных ChiP-Seq эксперимента. Для выполнения
задания я использовала следующую команду (параметр
--nomodel
введен, так как пиков было слишком мало):
$ macs2 callpeak -n chipseq_chunk41 -t chipseq_chunk41_sorted.bam --nomodel
При этом было получено 3 файла (
*.narrowPeak,
*.xls,
*.bed) с информацией
о пиках. Всего было найдено 11 пиков на 22 хромосоме (табл. 2).
Визуализация пиков в
UCSC Genome Browser
представлена на рис. 5. Для визуализации в браузер подавался
файл
chipseq_chunk41_peaks.narrowPeak,
в начало которого было дописано
"track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 41"
browser position chr22:28762900-29493050".
Таблица 2. Информация о найденных пиках
| № |
Начало |
Конец |
Длина |
Расстояние от старта
пика до его вершины |
-log10(p-value) |
| 1 |
28762906 |
28763274 |
369 |
187 |
33.06771 |
| 2 |
28820739 |
28821101 |
363 |
130 |
23.34650 |
| 3 |
29179854 |
29180076 |
223 |
47 |
14.57349 |
| 4 |
29204765 |
29205243 |
479 |
123 |
23.51211 |
| 5 |
29215520 |
29215520 |
361 |
161 |
28.35736 |
| 6 |
29225808 |
29226007 |
200 |
38 |
11.12255 |
| 7 |
29288442 |
29288643 |
202 |
117 |
17.59388 |
| 8 |
29307871 |
29308181 |
311 |
184 |
17.91896 |
| 9 |
29319981 |
29320249 |
269 |
150 |
15.97544 |
| 10 |
29378099 |
29378298 |
200 |
30 |
15.28724 |
| 11 |
29492807 |
29493042 |
236 |
76 |
12.89292 |
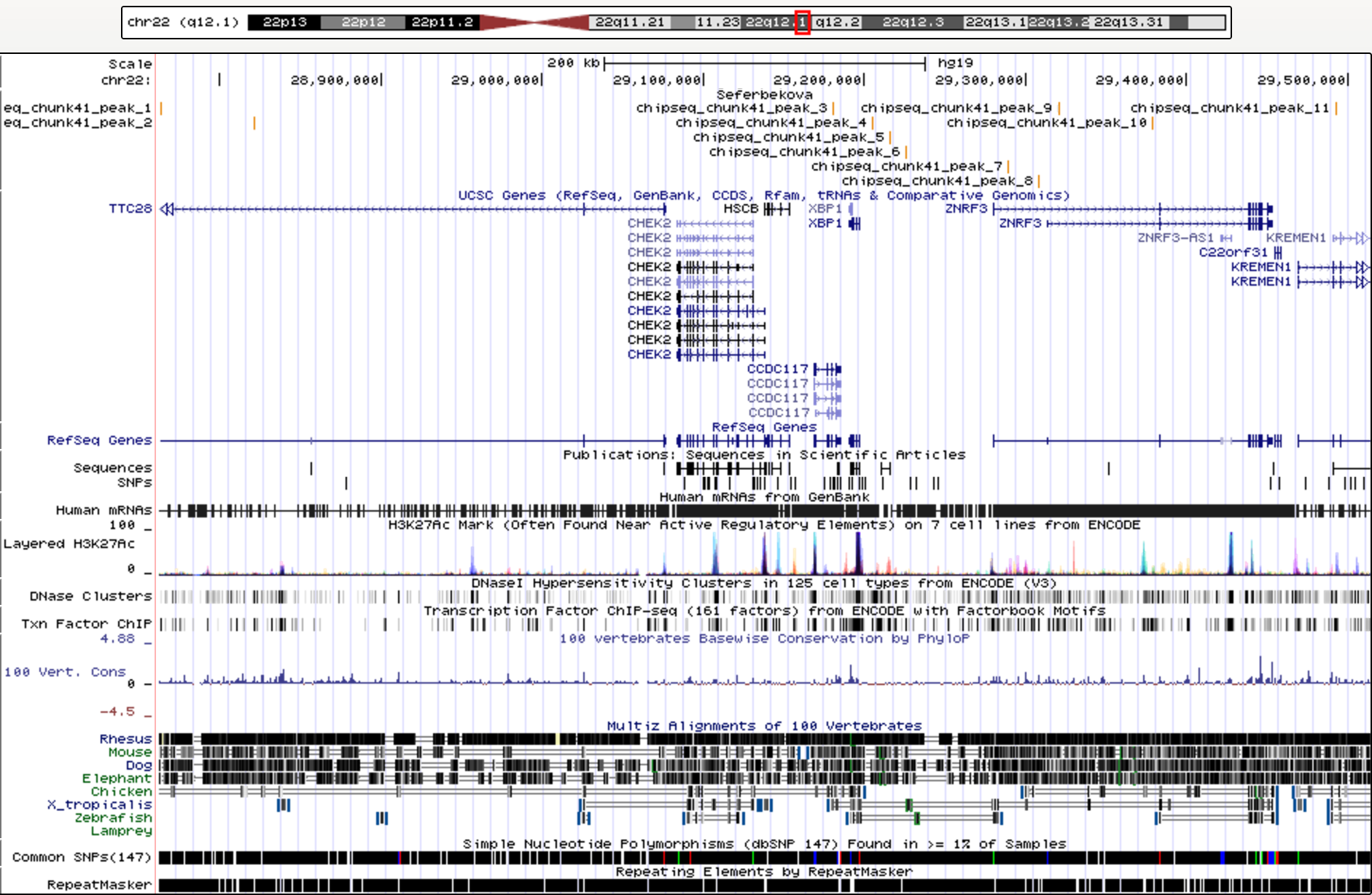
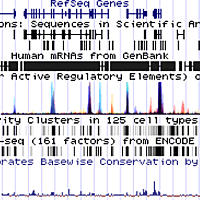
Рис. 5. Визуализация пиков в UCSC Genome Browser
|
Каждому найденному пику соответствует определенное число, позволяющее
оценить достоверность находки, — -log
10(p-value).
При этом чем больше это число, тем меньше p-value и, следовательно,
тем выше достоверность находки. В принципе, у всех находок достаточно
низкий p-value. Тем не менее я решила рассмотреть первые две, как
наиболее статистически достоверные. На рис. 6 и 7 представлены
изображения двух этих пиков в
UCSC Genome Browser.
Рис. 6. Визуализация первого пика в UCSC Genome Browser
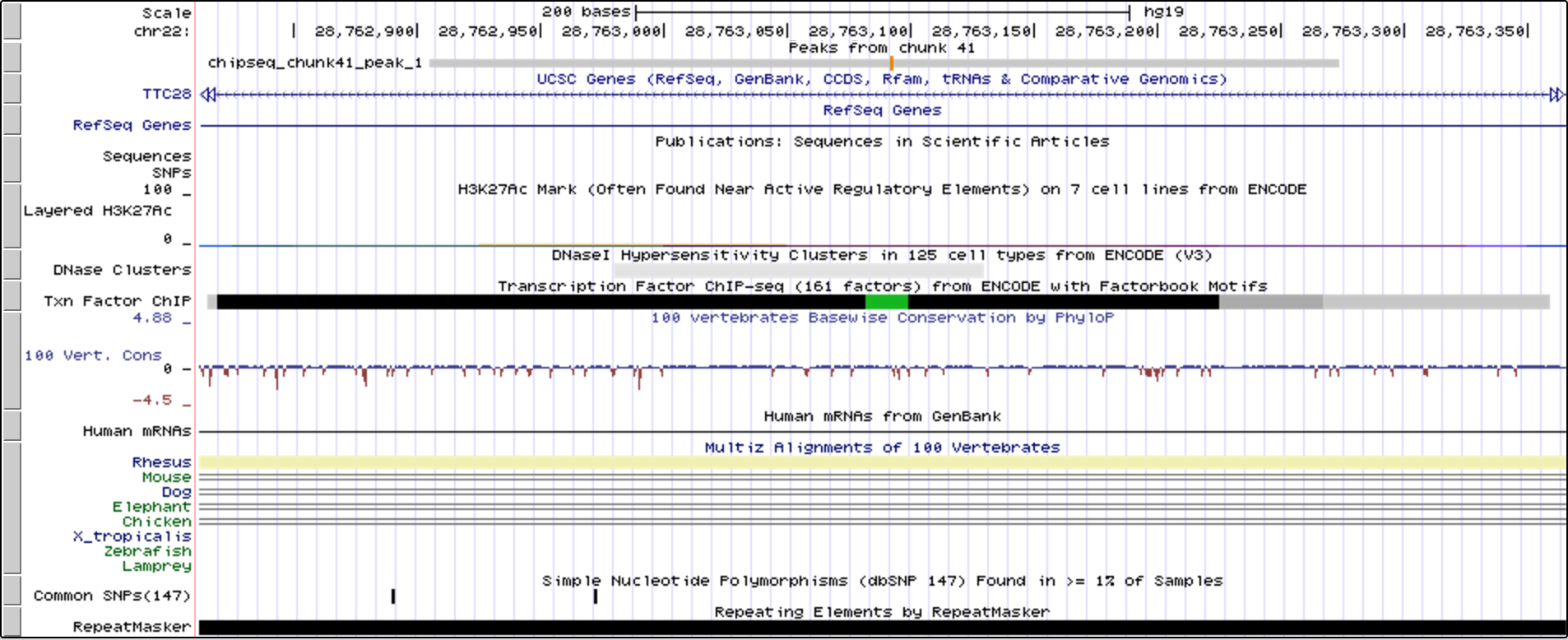
Рис. 7. Визуализация второго пика в UCSC Genome Browser
Как можно заметить, оба пика попали на ген
TTC28, а точнее на его интроны. Этот ген кодирует белок с
тетратрикопептидным мотивом, который во время митоза участвует в
конденсации микротрубочек в центре веретена и образовании тельца
Флемминга. Тем не менее, так как пик целиком попадает на интрон,
влияние возможного связывания ТФ с этим участком вряд ли значимо.
Далее при помощи команды:
/srv/databases/ngs/tools/seqtk/seqtk subseq /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk41_peaks.narrowPeak
> my_peaks.fa
был получен файл в формате
.fasta,
последовательности из которого затем были выровнены в программе
Jalview (Muscle with defaults). Полученное выравнивание
представлено на рис. 8.
Рис. 8. Выравнивание последовательностей (окраска ClustalX)
Как видно на изображении, в выравнивании очень много гэпов и мало
консервативных позиций. Определить консенсусную последовательность
длиной 4-8 нуклеотидов и с хотя бы 70% консенсуса для каждой позиции
не представляется возможным.