Реконструкция эволюции доменной архитектуры
Выбор домена и его описание
Для выполнения задания я выбрала домен
ZZ zinc finger
(ID: ZZ, AC: PF00569). Этот домен найден у
786 видов в составе
6142 последовательностей белков, и всего для него известно
536
архитектур.
Выбранный домен является одним из типов
цинковых
пальцев (анг. "zinc finger"). Домены этого типа связывают один или
несколько ионов цинка посредством координационных связей с
аминокислотами (обычно это 2 гистидина и 2 цистеина)
1.
Белки, содержащие цинковый палец, в большинстве своем связываются
с ДНК, РНК, другими белками или небольшими молекулами.
Конкретно мой домен содержит 4-6 остатков цистеина, которые связывают
два иона цинка
2. Этот домен также содержит мотив
Cys-X2-Cys, найденный в других цинковых пальцах. Считается,
что цинковый палец типа ZZ участвует в белок-белковых взаимодействиях:
к примеру, он был найден в таких белках, как Е3 убиквитин-лигаза и
дистрофин.
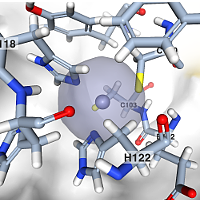
На рис. 1 представлено изображение домена в составе Е3 убиквитин-лигазы
MIB1 человека (PDB ID:
4XI6), а на рис. 2 и 3 — приближенное изображение
взаимодействия двух координационных центров с ионами цинка
(фиолетовые).
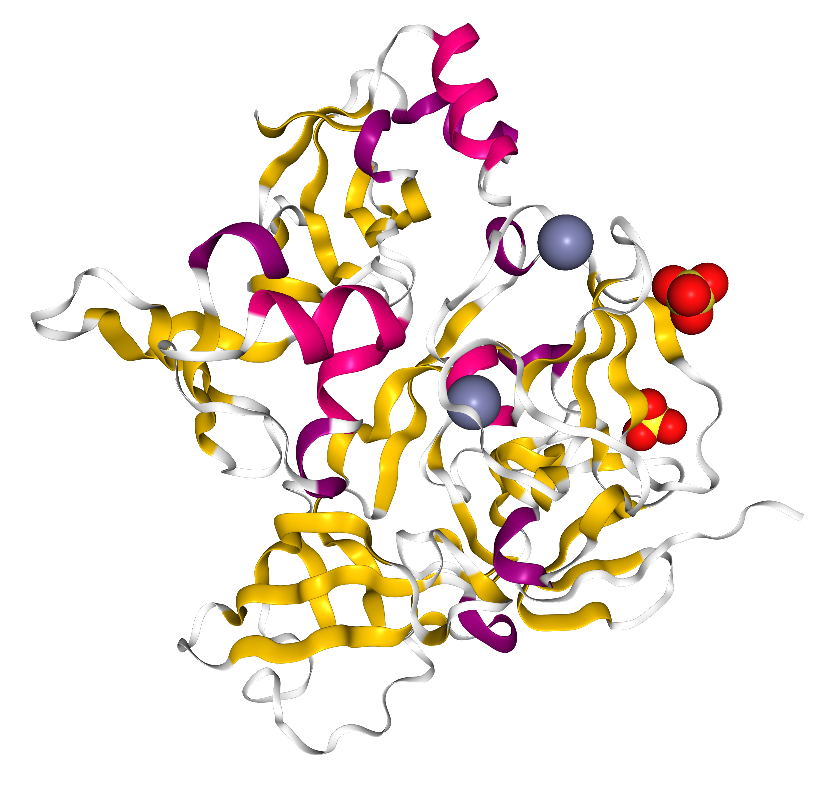
Рис. 1. Визуализация
домена ZZ |
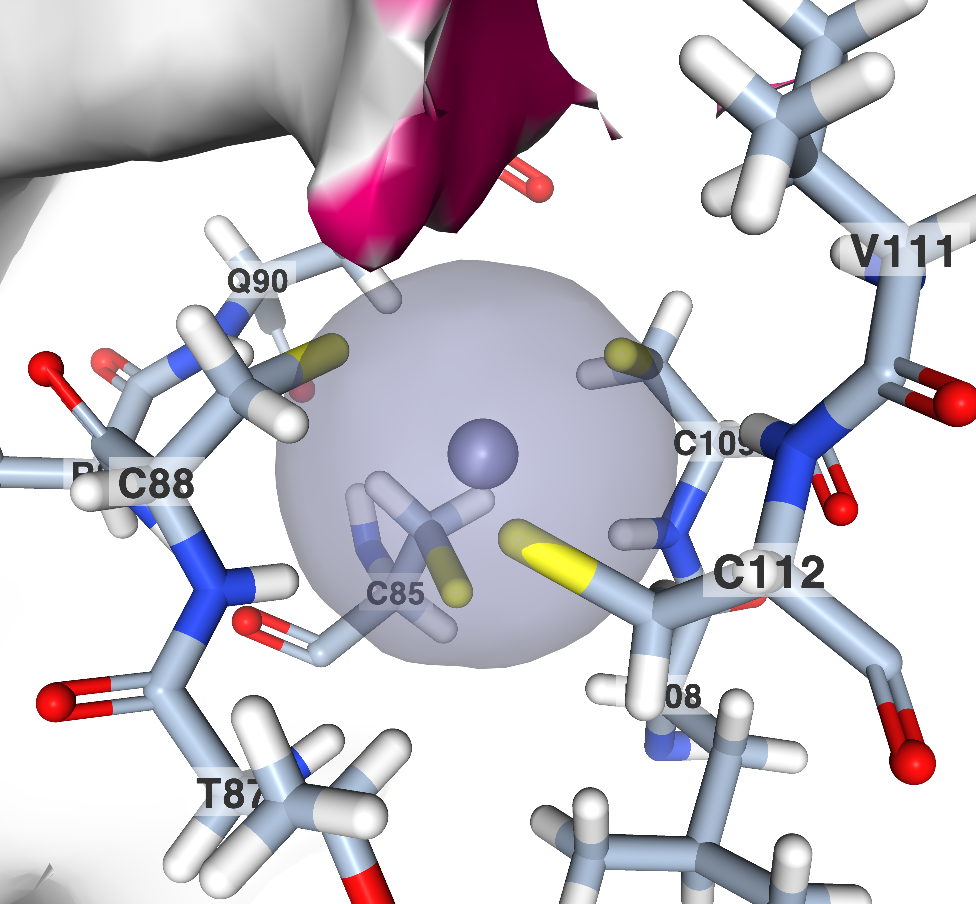 Рис. 2.
Координационные связи домена с цинком Рис. 2.
Координационные связи домена с цинком |
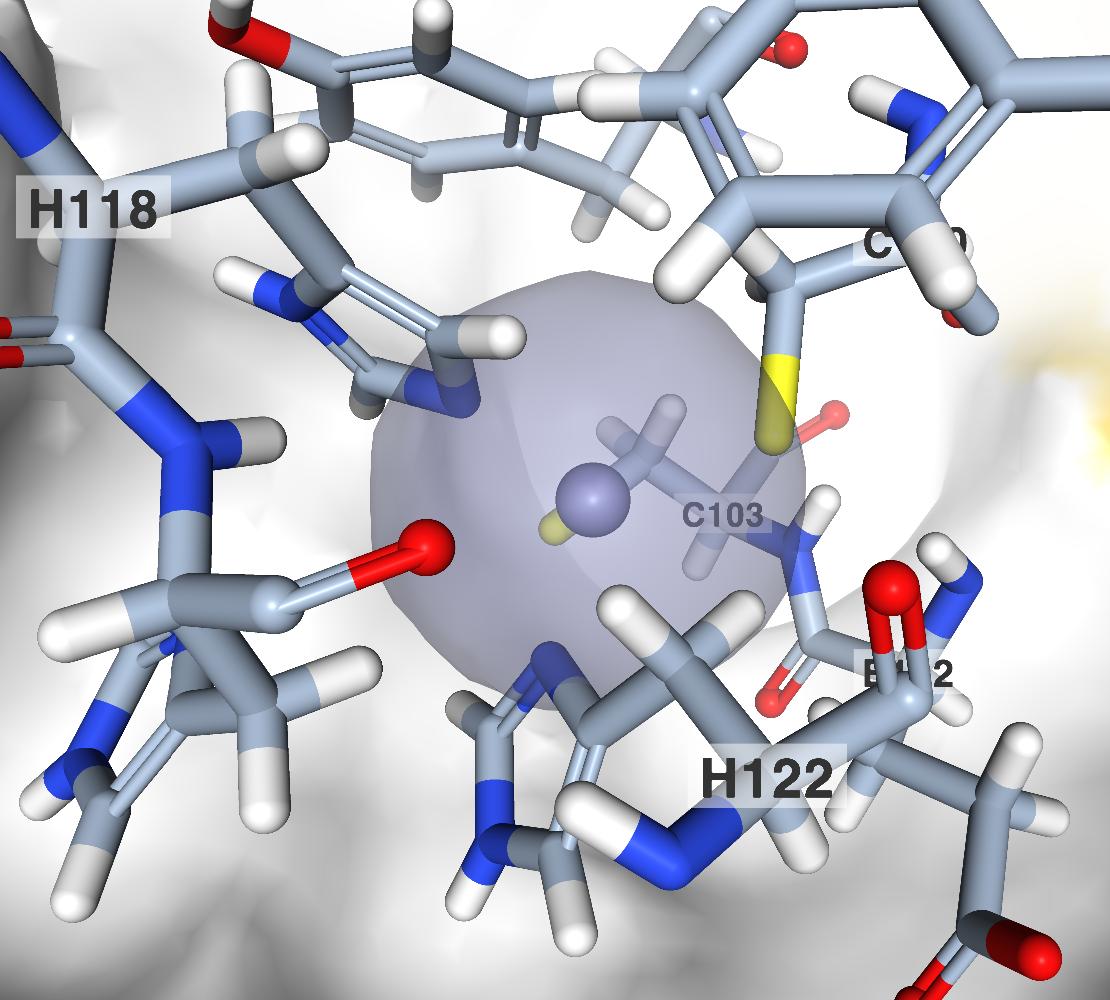 Рис. 3.
Координационные связи домена с цинком Рис. 3.
Координационные связи домена с цинком |
Построение выравнивания
Для выбранного домена было скачано выравнивание (
Jalview:
File →
Fetch Sequences → Pfam (Full) → PF00569), которое затем было
покрашено (
ClustalX, порог консервативности=30). Также я добавила
3D структуру белка
DTNA_HUMAN, последовательность которого выделена
в выравнивании оранжевым цветом. Полученное выравнивание доступно
в формате
.fasta или в качестве
проекта.
К сожалению,
Jalview отказался сохранять изображение выравнивания
(оно слишком большое), поэтому привести его здесь я не могу. Отмечу только,
что в выравнивании очень много гэпов и очень мало консервативных позиций,
не говоря о блоках. Это можно связать с большим числом последовательностей.
Выбор архитектур и их описание
Для моего домена известно всего 536 архитектур
(
пруф). Из
них я выбрала вторую и третью архитектуры: {EF-hand_2, EF-hand_3, ZZ} и
{ZZ, Myb_DNA-binding}. Они найдены в 507 и 349 последовательностях
соответственно и представлены на рис. 4 и 5.

Рис. 4. Архитектура {EF-hand_2, EF-hand_3, ZZ},
далее обозначаемая I.
Красный — EF-hand_2,
синий — EF-hand_3, зеленый —ZZ.
 Рис. 5.
Архитектура {ZZ, Myb_DNA-binding}, далее обозначаемая II. Рис. 5.
Архитектура {ZZ, Myb_DNA-binding}, далее обозначаемая II.
Зеленый —ZZ, желтый — Myb_DNA-binding. |
|
Сначала с помощью скрипта
swisspfam_to_xls.py я отобрала последовательности
с моим доменом из файла
/srv/databases/pfam/swisspfam.gz, содержащего
информацию об архитектуре всех последовательностей:
python swisspfam-to-xls.py -z -i swisspfam.gz -p PF00569 -o archs.xls
Для полученной таблицы была составлена сводная таблица, в которой столбцами
являются разные домены, а строками — идентификаторы белков. Исследуемый
домен выделен красным, два белка из I — синим, а из II — зеленым.
Затем для всех идентификаторов были скачаны соответствующие последовательности
(
Uniprot → Retrieve), для которых была получена таксономия с помощью
скрипта
uniprot_to_taxonomy.py:
python uniprot-to-taxonomy.py -i uniprot.txt -o tax.xls
Полученная таблица с таксономией была добавлена к предыдущей. При этом
я выбрала в качестве таксона было выбрано царство Metazoa (животные),
а в качестве двух подтаксонов — Ecdysozoa или
Линяющие
(далее
Е) и Chordata или
Хордовые (далее
С).
Соответственно в столбце таксономии указаны царства и типы.
Затем с помощью небольшого
скрипта я получила
длины мотивов и вставила их в общую таблицу с помощью ВПР. Таким образом,
была составлена таблица, представленная на листе
architectures_selected,
на котором выписана собранная информация о всех последовательностях. Из них
было выбрано по 30 последовательностей для каждого из двух таксонов. Они
отмечены "+" в колонке "selected" на упомянутом листе. Итоговая таблица:
скачать.
Сохранив отдельно
файл с нужной информацией, я получила
два файла:
ids_tax.txt с модифицированными
идентификаторами и
ids.txt с исходными идентификаторами
при помощи несложного
скрипта.
Затем с помощью скрипта
filter-alignment.py я оставила в выравнивании
только выбранные мною последовательности:
python filter-alignment.py -i align.fa -m ids.txt -o align_selected.fa -a "_"
Но и тут меня ждал подвох: в выравнивании осталось лишь 115 последовательностей
из 120, то есть 5 последовательностей не были отобраны скриптом. Я нашла
эти последовательности с помощью другого несложного скрипта и оказалось,
что это
Q9Y4J8, A2CI98, A2CJ06, Q8IYH5. Их идентификаторы
в выравнивании отличались:
DTNA_HUMAN, DYTN_MOUSE, DYTN_HUMAN, ZZZ3_HUMAN
соответственно. Поэтому их мне пришлось вернуть в выравнивание
вручную (спасибо, что их было всего лишь 4).
К идентификаторам в полученном выравнивании с помощью
скрипта я приписала выбранные обозначения для таксонов и архитектур: E1 и
E2 для линяющих и C1 и C2 для хордовых в соответствии с архитектурой.
соответствующие обозначения архитектур и таксонов. Итоговое выравнивание
с отобранными последовательностями:
align_selected.fasta.
Анализ выравнивания и дерева
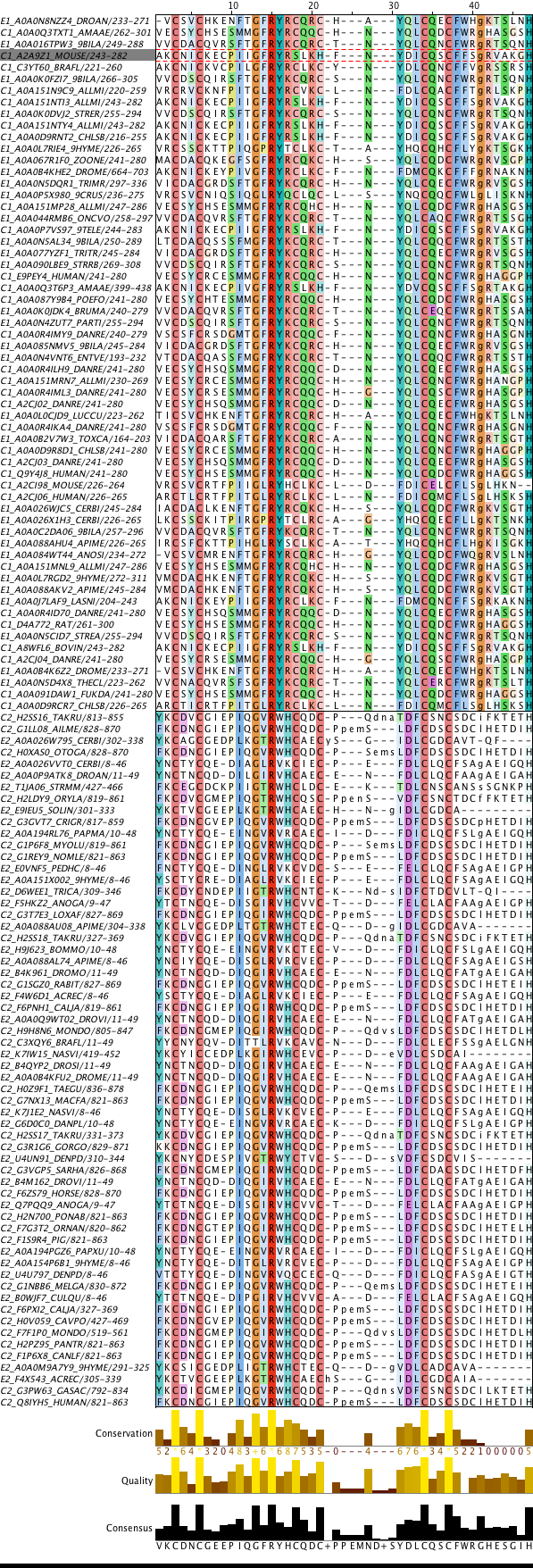
После всех мучений полученный файл с выравниванием доменов выбранных
последовательностей был открыт в
Jalview, и я провела чистку выравнивания: были
удалены пустые колонки, N- и C-участки; а потом создала две группы для
архитектур и раскрасила их
ClustalX (20%). Хочу отметить, что удалять
последовательности я не стала, так как большая часть отличий характерна
последовательностям E2. На рис. 6 представлено
полученное выравнивание. Оно также доступно в виде
проекта.
На изображении выравнивания видно, что для первой архитектуры последовательности
домена содержат намного больше консервативных между таксонами позиций (и меньше
гэпов, так как почти все гэпы обусловлены инделями в последовательностях для второй
архитектуры). В целом, в выравнивании последовательностей со первой архитектурой
можно выделить целые довольно большие вертикальные блоки, в то время как в
последовательностях со второй архитектурой наблюдаются большие различия. Хочу
также отметить, что для второй архитектуры у последовательностей значительно
варьируются последние ~10 аминокислот (причем без привязки к таксону), которые
консервативны у последовательностей со первой архитектурой. Зато с началом
домена все в точности наоборот: у последовательностей со первой архитектурой
первые 2 позиции в выравнивании не консервативны, а у последовательностей с
второй архитектурой в первой позиции большое число сходных аминокислот
(отличия, опять же, связать с таксономией не выйдет).
Вообще говоря, я бы сказала, что в обоих случаях вертикальные блоки присутствуют.
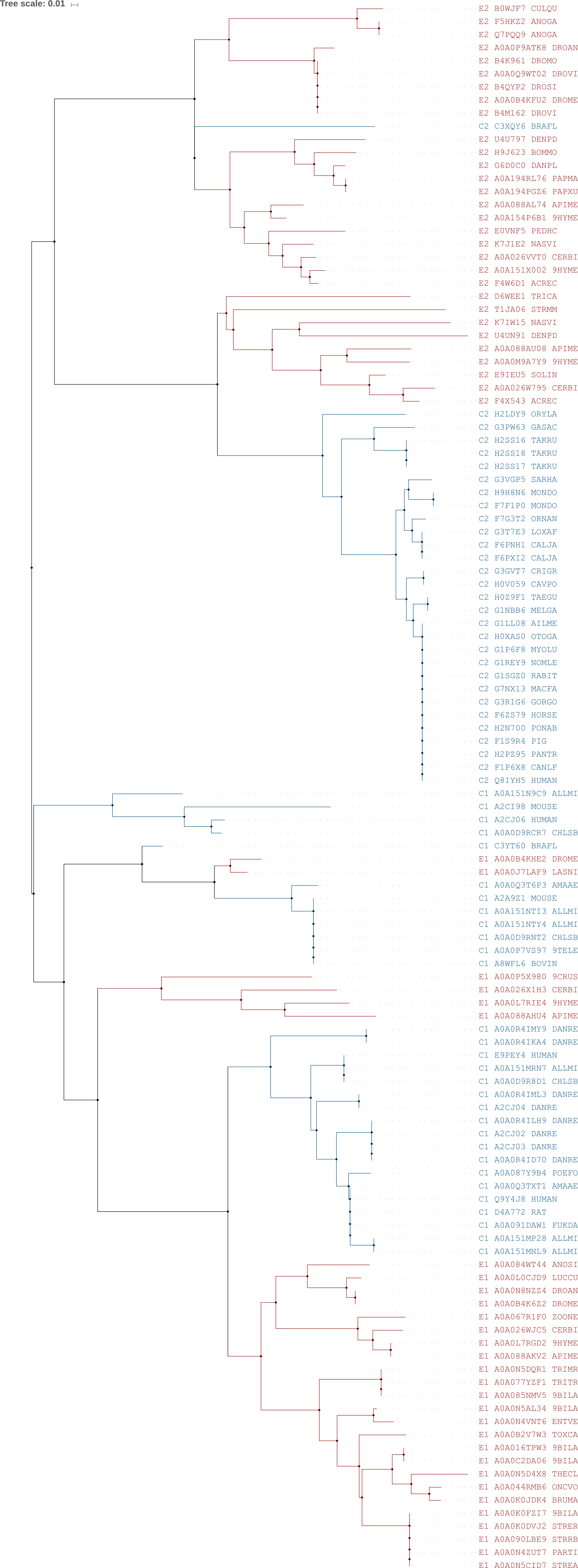
Для полученного итогового
выравнивания было
построено дерево с помощью программы
MEGA методом Neighbour-joining с
использованием bootstrap (100 реплик). Полученные деревья были сохранены в
формате
.nwk:
дерево с длинами ветвей,
дерево с бутстрэп-поддержкой ветвей. Построенное
дерево было укоренено в ветвь, разделяющую последовательности по архитектуре.
Затем я решила укоренить дерево в среднюю точку. Для этого
файл с деревом был подан на вход программе
retree и переукоренен
в среднюю точку. Таким образом, я получила
дерево с
тем же корнем: оно представлено на рис. 7. Полученное дерево также доступно в
классическом виде.
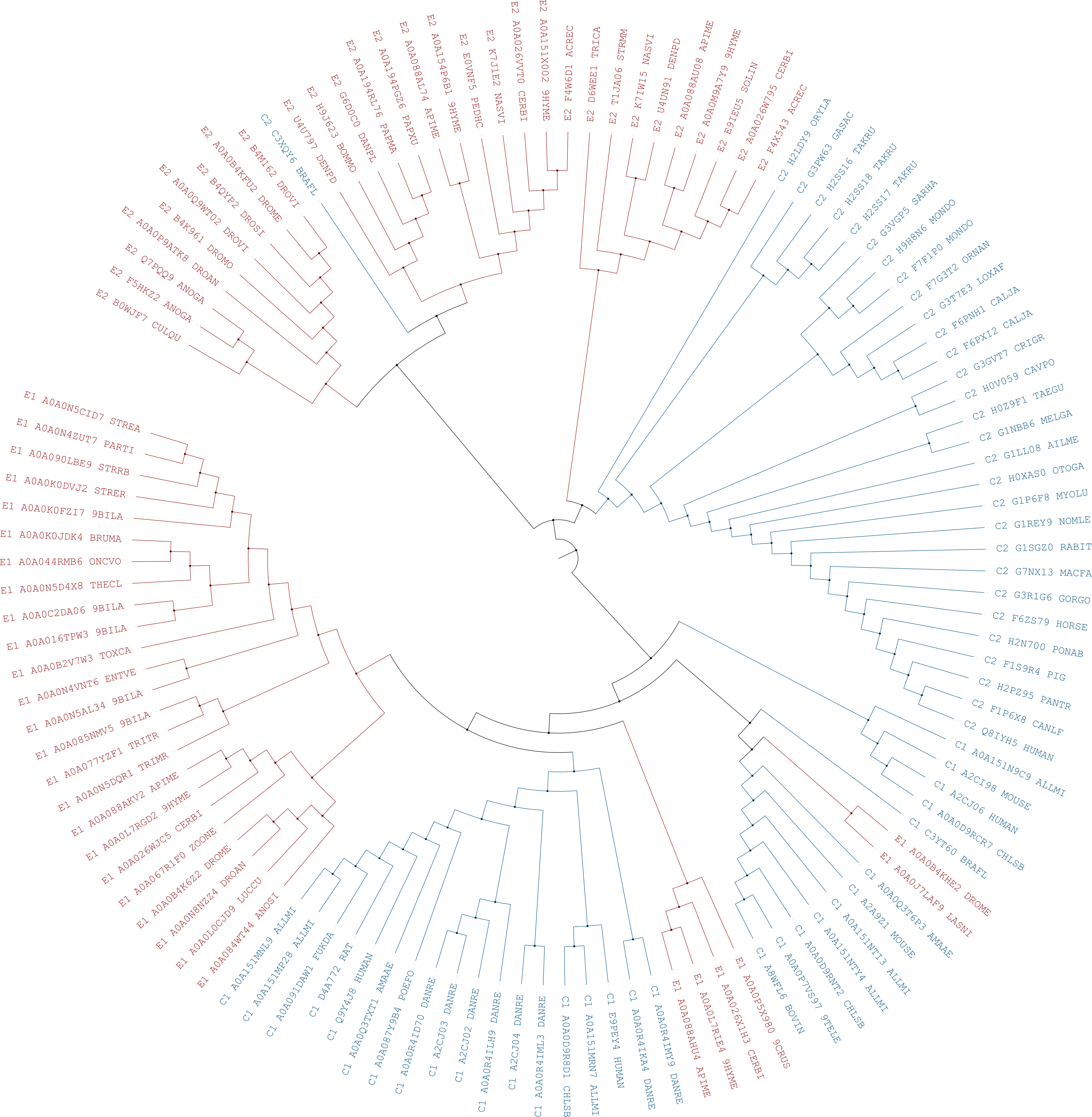 Рис. 7. Построенное методом NJ + bootstrap (100 реплик)
дерево. Красным выделены последовательности, относящиеся к линяющим;
синим — к хордовым. Дерево отражает только топологию; длины ветвей
учтены на дереве в классическом виде (см. выше)
Рис. 7. Построенное методом NJ + bootstrap (100 реплик)
дерево. Красным выделены последовательности, относящиеся к линяющим;
синим — к хордовым. Дерево отражает только топологию; длины ветвей
учтены на дереве в классическом виде (см. выше)
Как видно на рис.7, укоренение в среднюю точку привело к разделению дерева
на две клады, соответствующие двум архитектурам. Разделения по таксонам
четкого нет: "чужие" последовательности присутствуют в кладах другого таксона.
Таким образом, как мне кажется, у предкового организма уже существовало две
архитектуры для данного домена, которые затем эволюционировали в типах отдельно:
хорошо видно, что внутри клад по архитектурам последовательности собраны в клады
по типам, внутри которых последовательности сильно схожи.
Тем не менее, в некоторых кладах присутствуют последовательности из других
таксонов. Мне стало интересно, можно ли это каким-то образом объяснить.
К примеру, последовательность
C3XQY6_BRAFL — это домен
Branchiostoma floridae или хорошо известного всем ланцетника. Он
относится к типу хордовые, но на дереве последовательность домена из его
белка находится в кладе линяющих. Возможно, данный домен у ланцетника слабо
эволюционировал, в связи с чем его последовательность близка к последовательностям
линяющих из данной клады. Интересно также отметить, что для первой архитектуры
ветвь
C3YT60_BRAFL (тоже ланцетник) находится близко к ветвям
A0A0B4KHE2_DROME и
A0A0J7LAF9_LASNI, которые находятся в упомянутой
кладе с II архитектурой с последовательностью ланцетника. Получается, можно
проследить некоторую схожесть в эволюции этого домена у ланцетника и упомянутых
линяющих. С чем это может быть связано — сложный вопрос.
{kind=link}