Профили подсемейства белков
*Сурикаты иллюстрируют "семейство"
Выбoр подсемейства
Сначала необходимо было выбрать подсемейство из выравнивания
последовательностей домена, исследованного в
прошлом праке.
Подсемейство будем считать
хорошим, если оно является
кладой на дереве и если есть некоторые признаки (доменная архитектура,
таксономическая принадлежность или диагностические позиции), по которым можно
отнести домен к данному подсемейству.
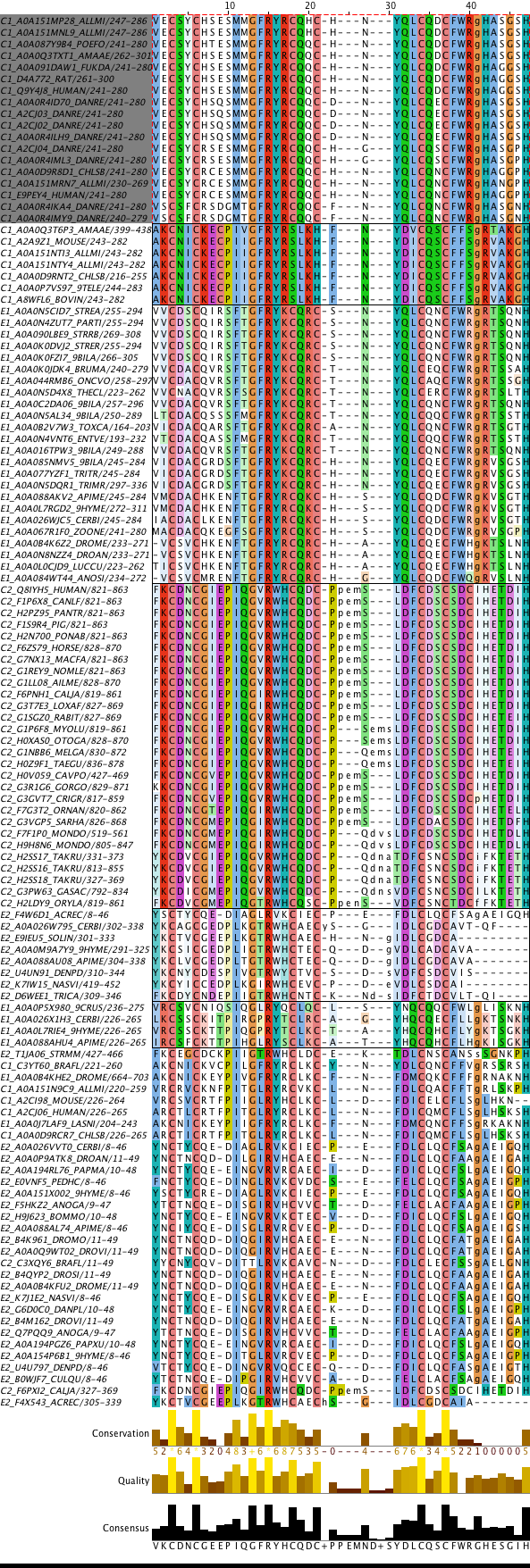
Я решила выбрать подсемейство, выделенное в выравнивании на рис. 1. Они
образуют кладу и имеют диагностические позиции (например, №1 и 4). Таким образом,
в подсемействе 18 последовательностей, а длина выравнивания последовательностей
их доменов 47. Выбранные последовательности были сохранены в отдельный файл в
формате
.fasta.
Построение профиля и поиск гомологов
Далее для полученного подсемейства необходимо было построить профиль. Для этого
использовался пакет
HMMER, использующий
скрытые модели Маркова для анализа выравнивания последовательностей и поиска
гомологов.
Сначала был построен профиль:
hmm2build profile.out domains.fa
Полученный файл c профилем был откалиброван:
hmm2calibrate profile.out
Затем я скачала с сайта
UniProt
полноразмерные последовательности белков, содержащих мой домен. По
полученному
файлу для итогового
профиля был осуществлен поиск гомологов:
hmm2search profile.out PF00569.fasta > findings.out
В итоговом файле
findings.out содержатся
все находки.
Анализ результатов
Для анализа результатов нам предложили использовать ROC-кривую и
гистограмму весов находок.
ROC-кривая (
Receiver Operator Characteristic)— кривая, которая
наиболее часто используется для анализа качества моделей. ROC-кривая
показывает зависимость количества верно классифицированных положительных
примеров от количества неверно классифицированных отрицательных примеров.
Рассмотрим возможные результаты:
- TP (True Positives) — верно классифицированные
положительные примеры (так называемые истинно положительные случаи).
В нашем случае — это количество последовательностей,
расположенных выше некоторого порога и достоверно содержащих искомый
домен;
- TN (True Negatives) — верно классифицированные
отрицательные примеры (истинно отрицательные случаи). В нашем случае
— это количество последовательностей, расположенных
ниже некоторого порога и достоверно не содержащих искомый домен;
- FP (False Positives) — отрицательные примеры,
классифицированные как положительные (ошибка II рода); Это
ложное обнаружение, т.к. при отсутствии события ошибочно выносится
решение о его присутствии (ложно положительные случаи). В нашем
случае — это количество последовательностей, расположенных
выше некоторого порога, но не содержащих домен;
- FN (False Negatives) — положительные примеры,
классифицированные как отрицательные (ошибка I рода). Это
так называемый "ложный пропуск" – когда интересующее нас событие
ошибочно не обнаруживается (ложно отрицательные примеры). В нашем
случае — это количество последовательностей, расположенных
ниже некоторого порога и содержащих домен.
Для самого анализа, однако, важны не эти абсолютные значения, а
относительные: специфичность и чувствительность.
Специфичность (
SP)
— доля истинно отрицательных случаев, которые были правильно идентифицированы
моделью (т.е. доля достоверно предсказанных белков, не содержащих домен, от общего
количества последовательностей, известно не содержащих этот домен).
Чувствительность
(
SE) — доля истинно положительных случаев (т.е. доля достоверно
предсказанных белков, содержащих домен, от общего количества последовательностей, известно
содержащих домен. Из определений следуют формулы, по которым считаются данные
показатели:
- SP = TN/(TN+FP)
- SE = TP/(TP+FN)
С использованием этих знаний, были проанализированы результаты поиска гомологов
по профилю. Для этого файл с находками был открыт в
Excel (лист 'profile').
Затем на другом листе ('roc') для каждого значения порога были подсчитаны шесть
описанных выше значения: TP, TN, FP и FN, а также SP и SN. Далее я также посчитала
значения 1-SP и построила график зависимости: по оси ОY откладывается SЕ, по оси ОX
— 1–SP (сто процентов минус специфичность). Полученный график и есть ROC-кривая.
Результат приведен на рис. 2. Итоговая таблица:
results.xlsx.
Далее я выбрала порог таким образом, чтобы SP и SE были одновременно максимальны. Это
то же самое, что и максимальное значение SP+SE-1. Таким образом, порог составил
105, при этом SP=0.963 и SE=1. Как видно, модель профиля позволяет с большой
достоверностью выделять подсемейство.
С помощью
Excel также была построена гистограмма весов находок. Необходимая
таблица и сама гистограмма находится на листе 'hist'. На рис. 3 представлен
полученный график.
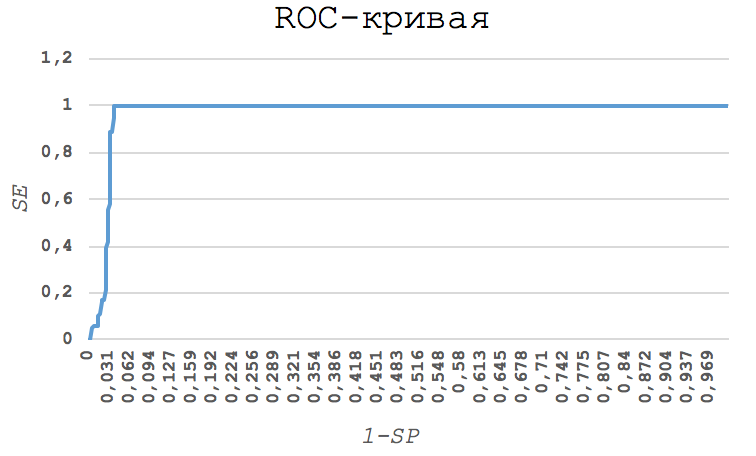
Рис. 2. Полученная ROC-кривая |
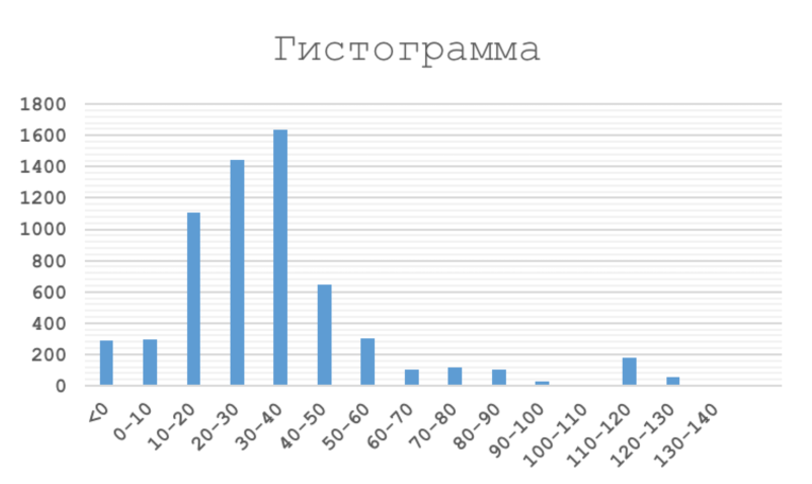
Рис. 3. Полученная гистограмма весов находок |
Таблица 1. Результаты поиска при выбранном пороге
| Находок |
принадлежит подсемейству |
не принадлежит подсемейству |
Сумма |
| выше порога по профилю |
18 |
220 |
238 |
| ниже порога |
0 |
5798 |
5798 |
| Сумма |
18 |
6018 |
6036 |
Поиск гомологов с помощью psi-BLAST