ЯМР vs РСА
Краткое описание методов
 [Если вы согласны с картинкой и просто шарите в теме,
этот раздел можно (и нужно) пропустить]
Спектроскопия ядерного магнитного резонанса
[Если вы согласны с картинкой и просто шарите в теме,
этот раздел можно (и нужно) пропустить]
Спектроскопия ядерного магнитного резонанса (ЯМР)
основана на поглощении электромагнитного излучения в области
частот 10 МГц - 1 ГГц (радиоволны) ядрами атомов, помещенных
в сильное внешнее магнитное поле (В
0). Магнитное
поле частично поляризует ядра, а сильное радиочастотное (РЧ)
поле возбуждает некоторые ядра. Когда радиочастотный сигнал
выключается, ядра возвращаются в исходное (более низкое)
энергетическое состояние, излучая небольшое количество
энергии. Это излучение фиксируется детектором, усиливается,
подвергается преобразованию Фурье, и таким образом получают
сигнал ЯМР — так называемый «химический сдвиг». Недостатком
ЯМР-спектроскопии является низкая чувствительность, обусловленная
самой методикой и лежащей в ее основе квантовой физикой, из-за
чего образец должен быть очень чистым и концентрированным.
Рентгеноструктурный анализ (РСА) основан на использовании
высокоэнергетических рентгеновских лучей (λ ~ 0.1 нм) для анализа
структуры белка. Так как размеры атомов имеют примерно тот же
порядок, что и длина волны излучения, эта волна отклоняется и
рассеивается электронами, окружающими атомы в кристалле белка.
Отклоненные лучи интерферируют и создают диффракционную картину.
Для усиления сигнала и получения дифракционной картины
необходимо много молекул белка, организованных в виде
упорядоченной решетки, то есть кристалла.
Одним из основных отличий двух методик является то, что при РСА
белок находится в кристаллизированном состоянии, поэтому полученная
структура отражает одно состояние белка в определенной ориентации.
В случае ЯМР белок находится в растворе, следовательно подвижен,
поэтому структура ЯМР представляет собой набор рассчитанных
моделей с самой низкой энергией. По этой причине для РСА не важен
размер исследуемой молекулы: если белок может быть кристаллизован,
то его можно исследовать. Для ЯМР же размер имеет значение: в
случае крупных молекул время релаксации сильно увеличивается,
ухудшая качество сигнала и приводя к размытию пика. Тем не менее,
в отличие от РСА, ЯМР дает представление о подвижности белка в
растворе (1).
Белок, используемый для сравнения
Для работы был выбран белок HSPC034, чья структура была
расшифрована методом РСА (PDB ID:
1TVG) и
ЯМР (PDB ID:
1XPW).
Разрешение структуры РСА 1.6 Å, количество моделей в структуре
ЯМР 20. На рис.1 показано наложение структур, полученных обоими
методами: РСА структура показана розовым, голубым — одна
(слева) или все (справа) модели ЯМР структуры. Видно, что в целом
структуры похожи; сильные отличия видны в подвижных петлях и
еще небольшие отличия наблюдаются ближе к концам бета-стрэндов.
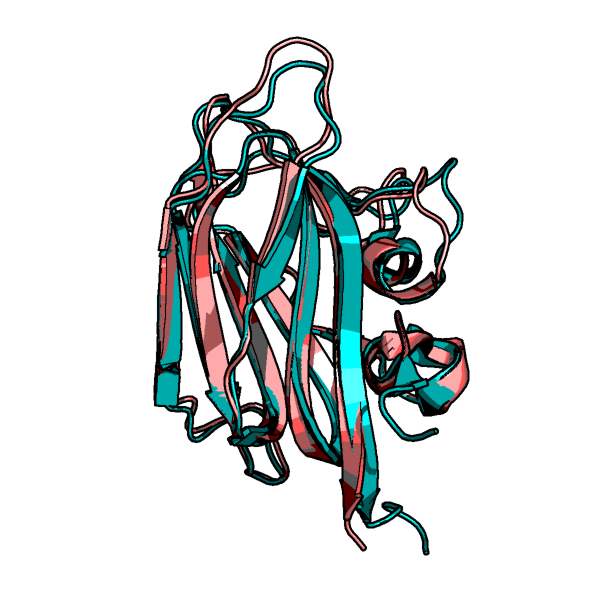
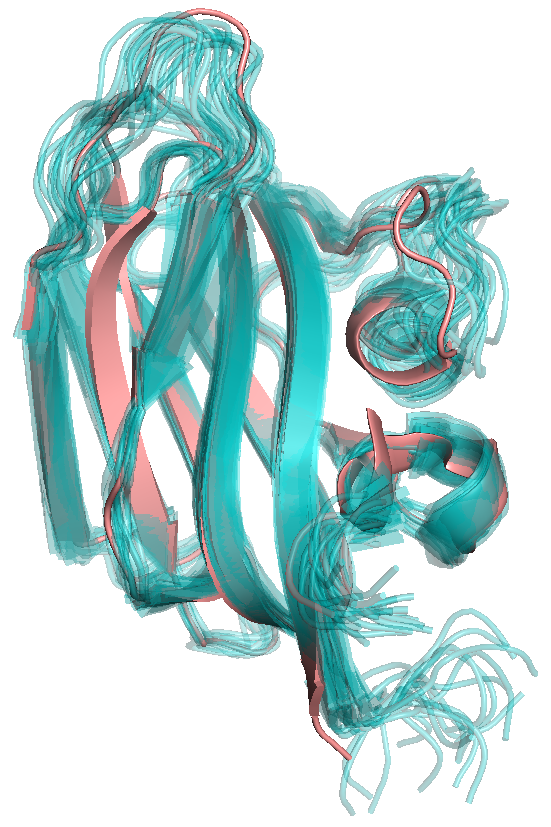
Рис.1. Наложение структур, полученных с помощью РСА
(розовый) и ЯМР (голубой) экспериментов. Для ЯМР слева
показана одна структура, а справа — все 20
Водородные связи
Будем считать, что между донором и акцептором протона есть
водородная связь, если расстояние между ними ≤ 3,5 Å. Для
изучения были выбраны три водородные связи: в бета-листе,
в ядре, в петлях на поверхности. Взаимодействующие остатки
показаны на рис. 2, 3, 4 соответственно, а в таблице 1 приведено
описание рассмотренных связей. Связей между боковыми цепями в
ядре белка найдено не было, поэтому я выбрала связь, более-менее
скрытую от выхода на поверхность другими остатками.
| № |
Остатки |
Описание |
Длина связи, Å
(РСА) |
Число моделей со связью
(ЯМР) |
Min длина связи, Å
(ЯМР) |
Max длина связи, Å
(ЯМР) |
Median длина связи, Å
(ЯМР) |
| 1 |
ILE64 (N)---(O) GLU103 |
Остов бета-листа |
2.84 |
20 (100%) |
2.80 |
3.20 |
2.95 |
| 2 |
GLN101 (NE2)---(OG) SER66 |
Боковые цепи ~ядра белка |
2.75 |
0 (0%) |
- |
- |
- |
| 3 |
HIS95 (NE2)---(OE1) GLU97 |
Боковые цепи петли на поверхности белка |
2.92 |
4 (20%) |
2.60 |
2.81 |
2.68 |
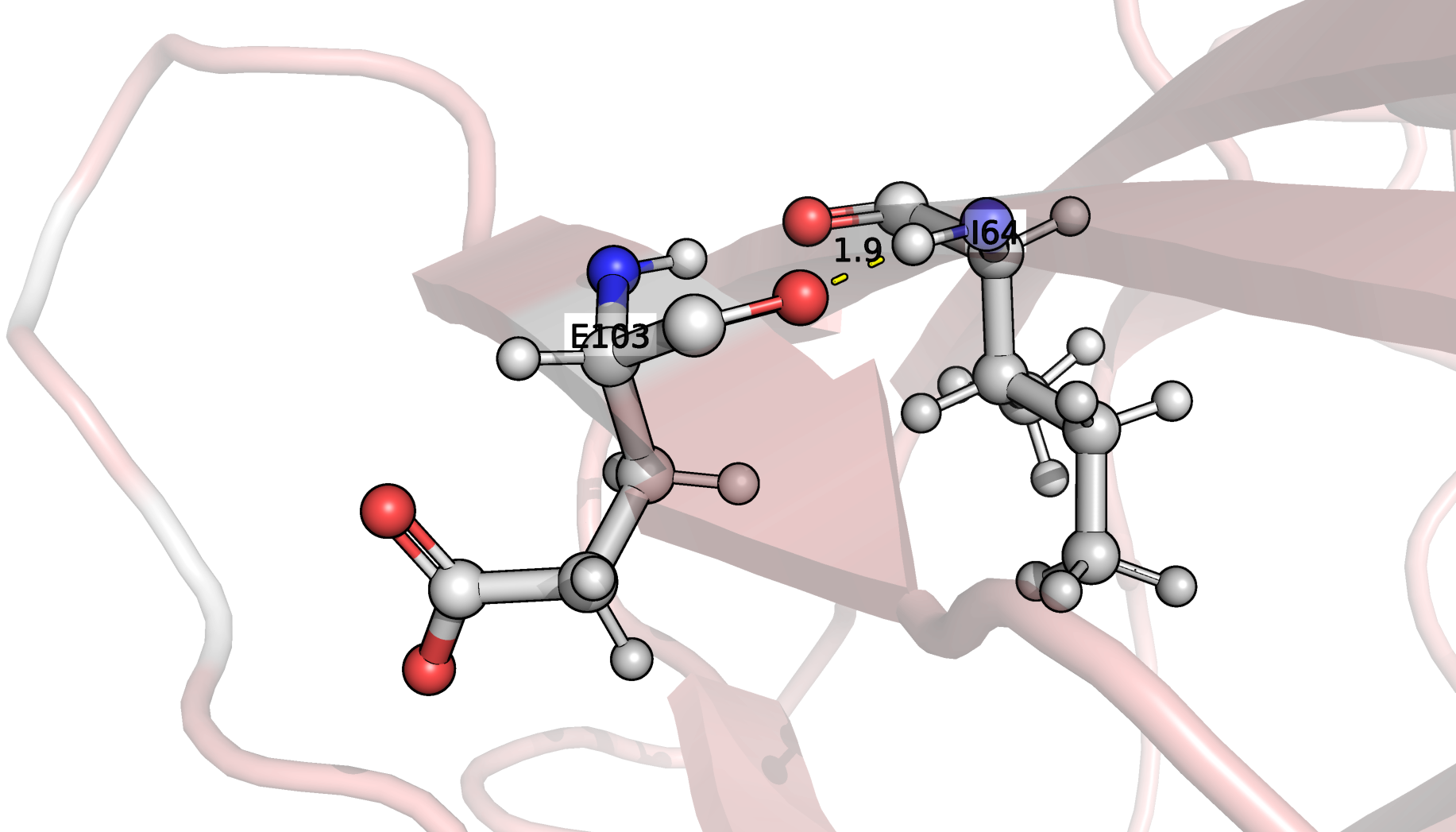
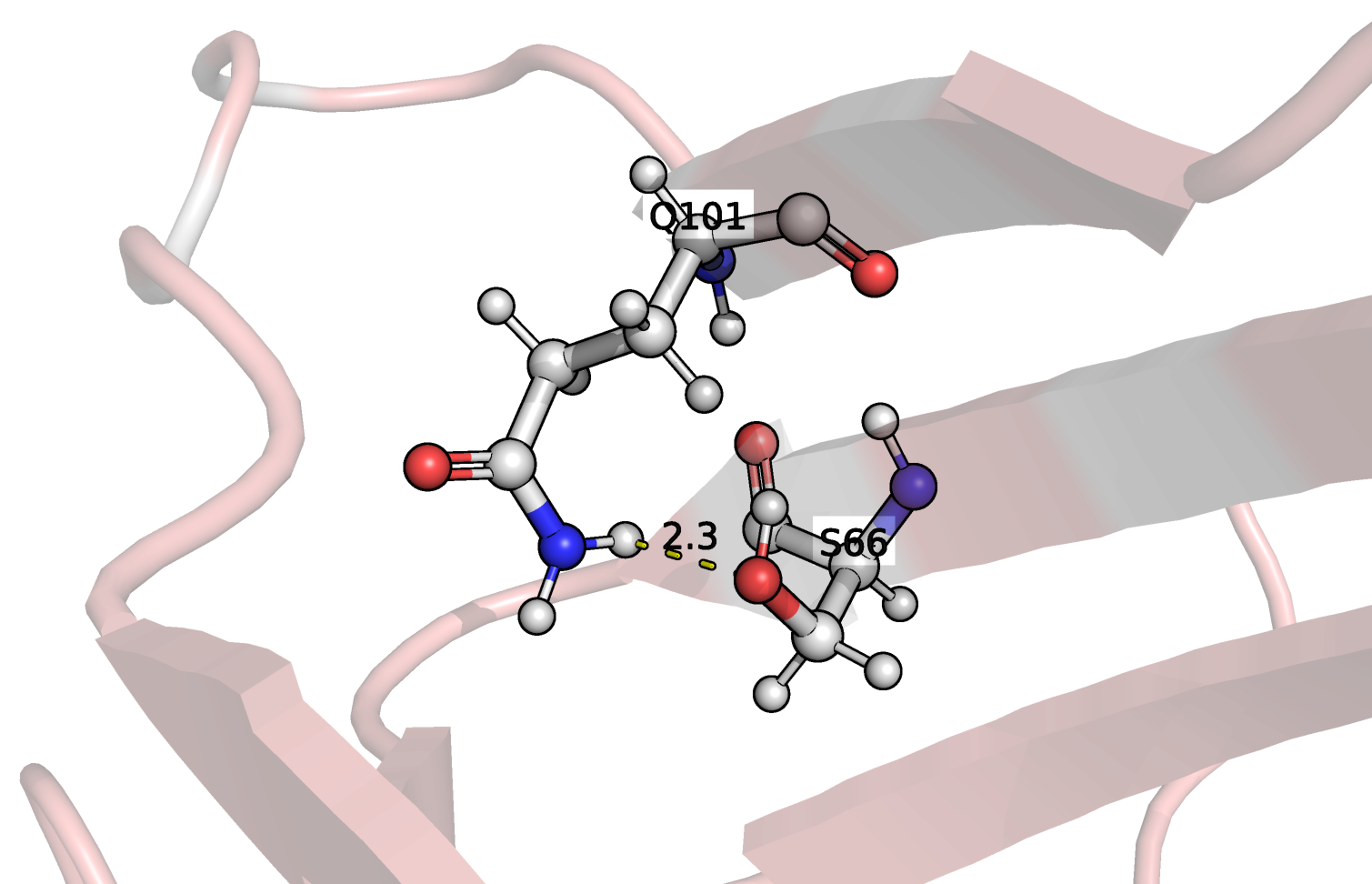
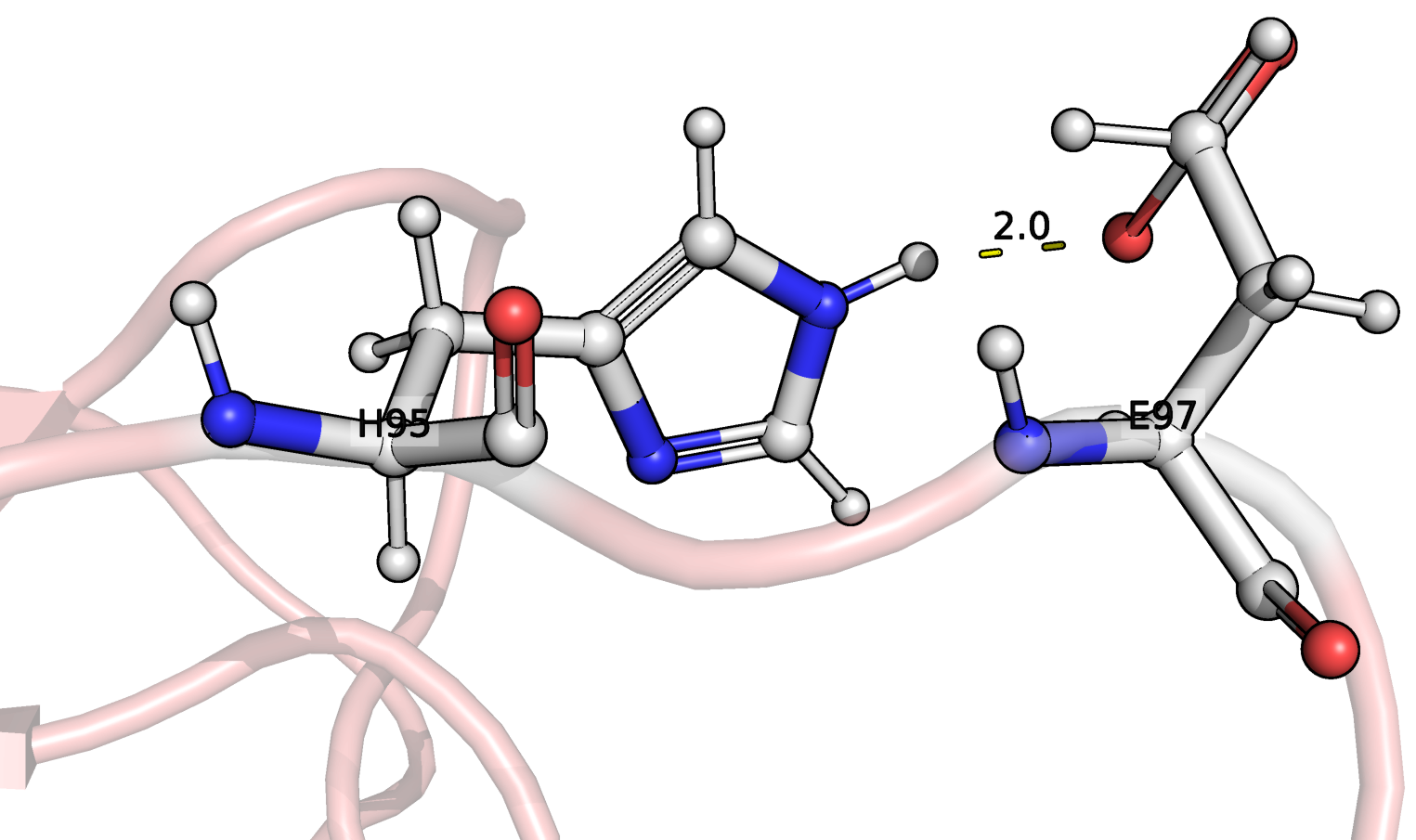


Рис.2. Рассматриваемые водородные связи, обсуждаемые
в тексте и описанные в таблице 1; (слева) связь
№1, (в центре) связь №2, (справа) связь №3.
Таким образом, во всех ЯМР моделях присутствуют связь #1,
локализованная в остове бета-листа. Связь #2 не обнаружена в
моделях ЯМР, что выглядит логично, так как она локализована
в очень подвижном конце одного из стрэндов. Связь #3, несмотря
на локализацию в петле, найдена в нескольких ЯМР моделях
все-таки была. Это связано с большей подвижностью петлей и
боковых цепей а.о. в принципе. Расстояния между взаимодействующими
атомами сравнимы в случае обоих типов структур.
Некоторые из использованных команд, если я (или заблудший(ая) сюда
недобросовестный(ая) студент(ка)) захочу повторить содеянное:
script.txt.
Ссылки
[1]
Battle Royale: NMR vs. X-ray Crystallography // LibreTexts
