Совмещение структур
Для работы использовался поиск по сходству структур в PDBeFold
для уже родной структуры 2zf5:
таблица с находками. Из
находок были выбраны структуры для 4 гомологов, для выбора
использовался порог по RMSD (от 0,8 до 2,5) и по длине
выравнивания (50% от длины белка). Выравнивание было построено
только для одной цепи белка глицерол киназы, так как это
гомодимер. .fasta файл с множественным
выравниванием структур доступен по
msa.fasta. Общие характеристики полученного выравнивания:
Number of aligned residues = 459, Overall RMSD = 1.091,
Number of aligned SSEs = 30O, verall Q-score = 0.3907. На рис.
1 показано само выравнивание, а на рис.2 — совмещение
структур.
Таблица 1. Выбранные структуры гомологов
| № |
Q-score |
RMSD |
NSSE |
Nalgn |
Nalgn / Nquery, % |
PDB ID |
| 1 |
0.8841 |
0.959 |
34 |
466 |
93.8 |
2dpn:B |
| 2 |
0.9032 |
0.990 |
37 |
454 |
91.3 |
3ezw:D |
| 3 |
0.8967 |
0.959 |
36 |
466 |
93,8 |
4e1j:B |
| 4 |
0.8801 |
1.007 |
36 |
470 |
94.6 |
3h45:X |
 Рис. 1. Полученное множественное структурное выравнивание
Рис. 1. Полученное множественное структурное выравнивание
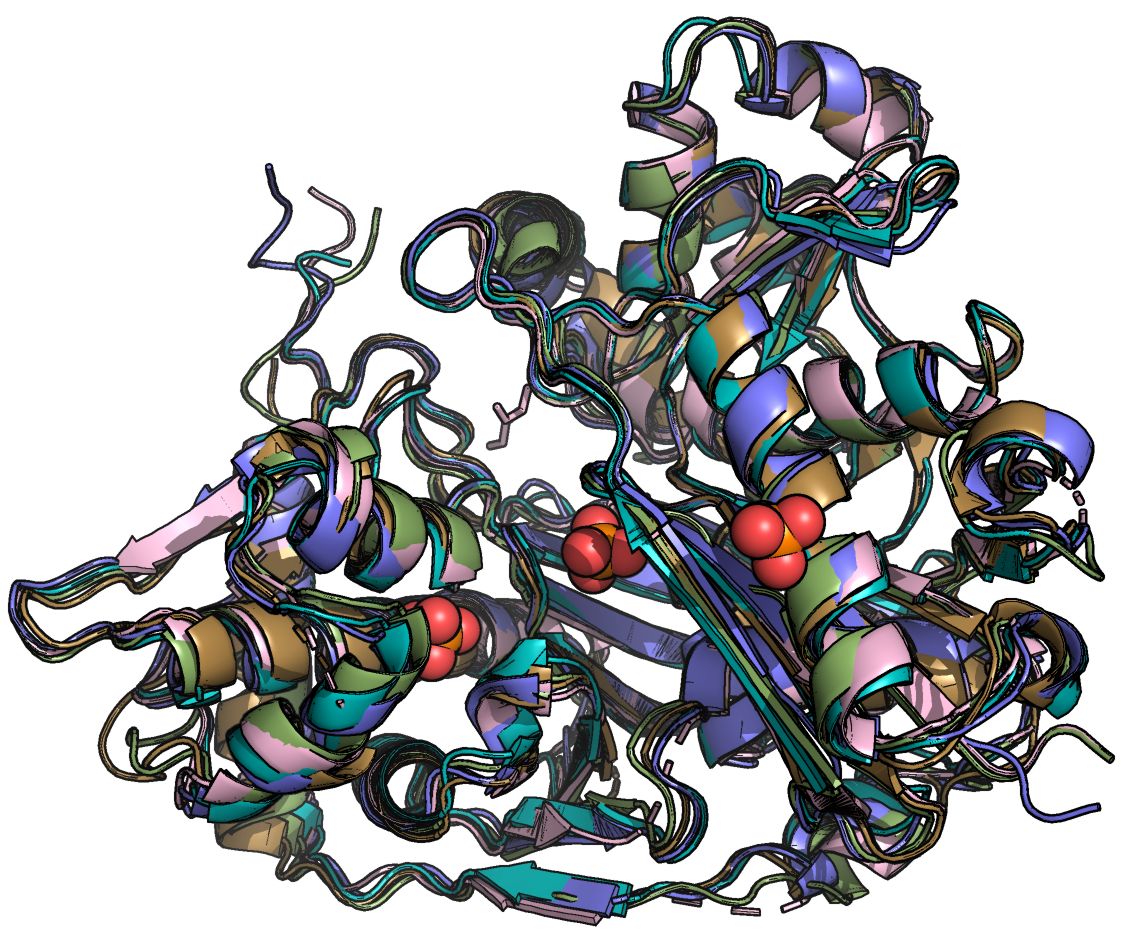 Рис. 2. Совмещение выровненных структур
Рис. 2. Совмещение выровненных структур
Далее с помощью
JalView было получено изображение структурного
выравнивания, показанное на рис. 3, а также было построено выравнивание
соответствующих белковых последовательностей алгоритмом
Muscle,
представленное на рис. 4 (результат в формате фаста:
msa_seq.fasta). Из выравнивания последовательностей была вырезана
одна из цепей (она не выравнялась).
Рис. 3. Полученное множественное структурное выравнивание
Рис. 4. Полученное множественное выравнивание последовательностей
Видно, что в целом оба выравнивания схожи. Отличается только положение
в выравниваниях нескольких остатков на границах структур: D91, E175,
E229, L230, 281-283...


