Главная I Семестр II Семестр III Семестр IV Семестр V Семестр VI Семестр Проекты
Официальный сайт ФББ Официальный сайт МГУ Полезные ссылки |
Докинг низкомолекулярных лигандов в структуру белка.
Цель работы - ознакомиться с возможностями докинга низкомолекулярного лиганда в структуру белка и пакета Autodock Vina и Autodock tools.
Работать будем с белком лизоцимом LYSC_HUMAN, структура которого (model5.pdb) была построена на основе гомологичного моделирования на прошлом занятии.
-
Программе Autodock Vina для докинга необходимы специально форматированные файлы pdb c зарядами и указанием торсионных углов. Для начала попробуем провести докинг одного из мономеров сахара (NAG) из прошлого занятия.
Для этого сначала найдем в банке pdb (www.pdb.org) SMILES аннотацию для NAG и сохраним ее в файле nag.smi.
-
Затем с помощью obgen построим 3D-структуру этого сахара в pdb-формате:
obgen nag.smi > nag.mol
babel -imol nag.mol -opdb nag.pdb
В результате был получен файл nag.pdb.
-
С помощью скрипта prepare_ligand4.py из пакета Autodock tools был создан pdbqt-файл лиганда NAG nag.pdbqt:
prepare_ligand4.py -l nag.pdb
-
С помощью того же скрипта был создан pdbqt-файл белка LYSC_HUMAN model5.pdbqt:
prepare_ligand4.py -l model5.pdb
-
Итак, мы получили входные файлы.
Для докинга необходимо указать область структуры белка, в которой будет происходить поиск места для связывания.
Удобно его задать как куб с неким центром. Координаты центра определим из модели комплекса, построенной на прошлом занятии.
Выберем атом сахара, находящийся в центре сайта связывания (например, атом C7B), и извлечем из текста pdb-файла model5.pdb его координаты (40.394, 40.690, 26.623).
Создадим по этим данным файл vina.cfg.
-
Проведем первый докинг:
vina --config vina.cfg --receptor model5.pdbqt --ligand nag.pdbqt --out nag_prot.pdbqt --log nag_prot.log
В результате докинга были получены файлы nag_prot.pdbqt и nag_prot.log.
-
Просмотрим файл nag_prot.log. Энергии трех лучших расположений и геометрическая разница между ними представлена в таблице:
| Расположение
| Энергия (ккал/моль)
| Геометрическая разница с лучшей моделью (rmsd l.b.)
|
| 1
| -5.9
| 0.000
|
| 2
| -5.6
| 2.004
|
| 3
| -5.5
| 1.690
|
Файлы nag_prot.pdbqt и model5.pdbqt были загружены в PyMOL.
Все состояния на одной картинке изображены ниже:
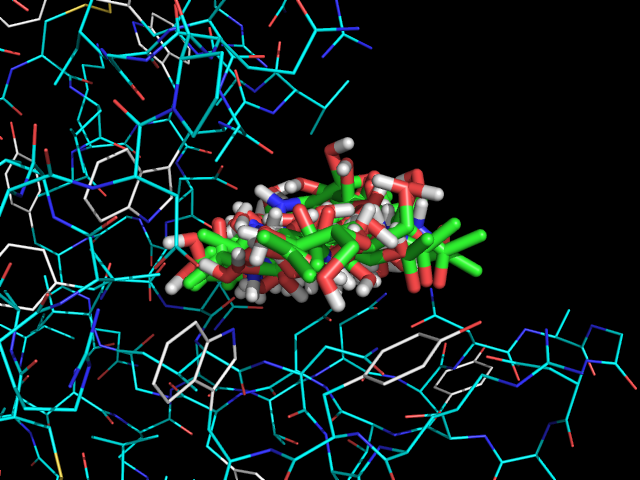
Такая картинка показывает, что молекула лиганда свободно перемещается внутри центра связывания белка.
Возможно, это связано с тем, что это лишь один из трех сахарных остатков реального лиганда.
-
Теперь проведем докинг, рассматривая подвижность некоторых боковых радикалов белка.
Сначала разобьем белок на две части: подвижную и неподвижную.
Для подвижной части выберем 3 аминокислоты, которые мы использовали в прошлом задании для позиционирования лиганда (Trp-82, Asp-120 и Glu-53).
Для создания pdbqt-файла воспользуемся скриптом prepare_flexreceptor4.py:
prepare_flexreceptor4.py -r model5.pdbqt -s GLU1_ASN5_ASP13
В результате были получены файл model5_flex.pdbqt и model5_rigid.pdbqt.
Теперь проведем докинг.
В результате получаем файлы vina_prot_flex.pdbqt и vina_prot_flex.log.
-
Просмотрим файл vina_prot_flex.log. Энергии трех лучших расположений и геометрическая разница между ними представлена в таблице:
| Расположение
| Энергия (ккал/моль)
| Геометрическая разница с лучшей моделью (rmsd l.b.)
|
| 1
| -5.1
| 0.000
|
| 2
| -4.9
| 1.409
|
| 3
| -4.9
| 1.337
|
Файлы model5_rigid.pdbqt и vina_prot_flex.pdbqt были загружены в PyMOL.
Все состояния на одной картинке изображены ниже:
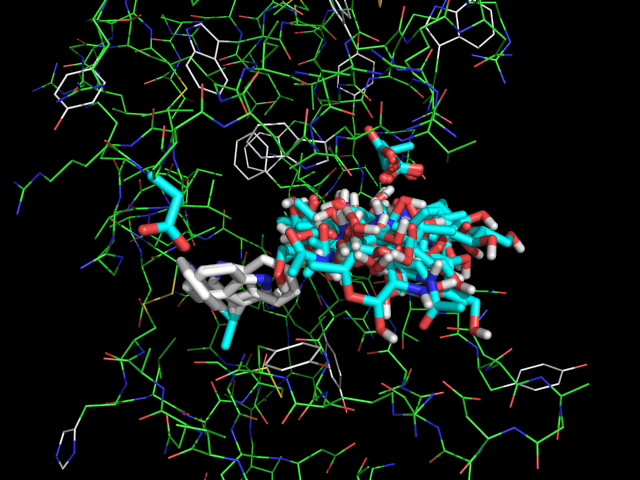
Как можно видеть, теперь свое положение меняет не только лиганд, но и три аминокислоты белка (правда, из них сильно изменяет положение только Trp-82; это связано с тем, что другие два остатка взаимодействовали в модели с другими мономерами лиганда).
На мой взгляд, такой подвижный докинг имеет куда больше биологического смысла, чем обычный докинг (ведь молекула белка не является жесткой неподвижной структурой).
Амплитуда перемещений лиганда в подвижном докинге куда больше, чем в обычном, что вполне естесственно объясняется подвижностью аминокислотных остатков. В некоторых состояниях очень хорошо видно, как Trp-82 сильно меняет конформацию, контактируя с лигандом.
-
К сожалению, докинг не смог расположить лиганд так, как он располагался в полученной модели.
Однако, стоит отметить, что лучше с задачей справился обычный докинг (а не подвижный!). Более того, только в обычном докинге образовалась водородная связь между лигандом и атомом азота остатка Trp-82 (причем, в двух моделях связь образовалась с нужным атомом кислорода лиганда).
Но по-настоящему хорошо с задачей не справился ни обычный, ни подвижный докинг.
-
Создадим три лиганда, где метильный радикал СH3C(=O)NH группы NAG заменен на OH, NH2 и H.
Для этого были получены файлы nag2.smi, nag3.smi и nag4.smi соответственно со SMILES измененных лигандов.
Затем были получены их pdb-файлы: nag2.pdb, nag3.pdb и nag4.pdb.
Наконец, с помощью скрипта prepare_ligand4.py были получены pdbqt-файлы: nag2.pdbqt, nag3.pdbqt и nag4.pdbqt.
В результате обычного докинга были получены файлы:
для первого лиганда: nag2_prot.log и nag2_prot.pdbqt;
для второго лиганда: nag3_prot.log и nag3_prot.pdbqt;
для третьего лиганда: nag4_prot.log и nag4_prot.pdbqt.
Таблица трех лучших расположений для -OH:
| Расположение
| Энергия (ккал/моль)
| Геометрическая разница с лучшей моделью (rmsd l.b.)
|
| 1
| -5.0
| 0.000
|
| 2
| -5.0
| 1.993
|
| 3
| -4.9
| 2.072
|
Для -NH2:
| Расположение
| Энергия (ккал/моль)
| Геометрическая разница с лучшей моделью (rmsd l.b.)
|
| 1
| -5.1
| 0.000
|
| 2
| -5.0
| 3.014
|
| 3
| -4.8
| 1.957
|
И для -H:
| Расположение
| Энергия (ккал/моль)
| Геометрическая разница с лучшей моделью (rmsd l.b.)
|
| 1
| -4.8
| 0.000
|
| 2
| -4.8
| 2.219
|
| 3
| -4.8
| 1.338
|
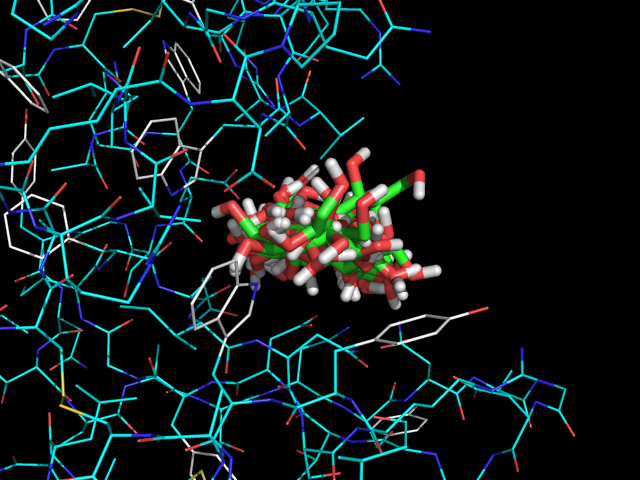
Изображение результата докинга для -OH:
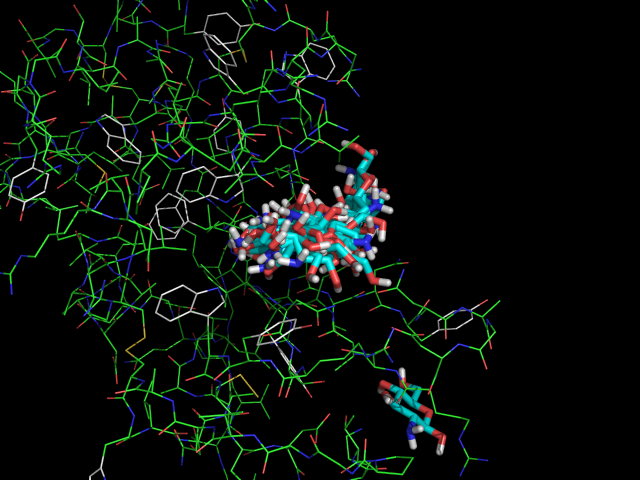
Изображение результата докинга для -NH2:
Изображение результата докинга для -H:
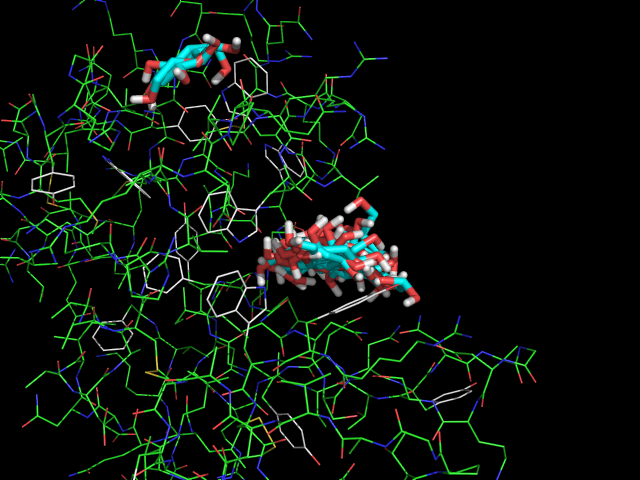
Интересно, что второй и третий лиганды в нескольких состояниях вышли за пределы центра связывания и оказались в совершенно другой части белка.
|