Полезные ссылки
Гомологичное моделирование комплекса белка с лигандом.
Цель работы - ознакомиться с возможностями гомологичного моделирования комплекса белка с лигандом и пакетом Modeller.Работать будем с человеческим лизоцимом (LYSC_HUMAN). Используя известную структуру лизоцима форели (запись PDB 1LMP) как образец, построим модель комплекса человеческого лизоцима с лигандом.
-
Построим выравнивание последовательностей белка 1lmp и LYSC_HUMAN с помощью программы ClustalW. Последовательность белка LYSC_HUMAN была сохранена в файле lysc_human.fasta.
Полученное выравнивание было сохранено в файле lysc_aligned.pir.
-
Модифицируем файл выравнивания. Во-первых, переименуем последовательности на ">P1;1lmp" и ">P1;seq".
Во-вторых, после имени последовательности моделируемого белка добавим строчку:
sequence:ХХХХХ::::::: 0.00: 0.00
Эта строчка описывает входные параметры последовательности для modeller. После имени последовательности белка-образца добавим строчку:structureX:1lmp_now.ent:1 :A: 130 :A:undefined:undefined:-1.00:-1.00
Эта строчка описывает, какой файл содержит структуру белка с этой последовательностью (1lmp_now.ent), номера первой и последней аминокислот в структуре, идентификатор цепи и т.д. В конце каждой последовательности добавим символы:/.
Этот символ означает конец цепи белка. Точка указывает на то, что имеется один лиганд. При этом оставляем строчку со звездочкой (*).
-
Модифицируем файл со структурой.
Удалим всю воду из структуры, всем атомам лиганда присвоим один и тот же номер "остатка" (130), одно и то же имя остатка (NAG), модифицируем имена атомов каждого остатка, добавив в конец буквы A, B и C (чтобы у каждого атома было разное имя).
То есть в результате этого атомы остатка 130 имеют индекс A, атомы остатка 131 - B, остатка 132 - C.
Сохраним изменения в файле 1lmp_now.ent.
-
Создадим управляющий скрипт lysc_human.py.
В нем указано:
-
что нужно использовать стандартные валентные углы в полипептидной цепи (строчка 4);
что дополнительно нужно сохранять взаимное расположение определенных пар атомов (3.5 ангстрема) - в данном случае трех атомов белка, образующих водородные связи с тремя атомами лиганда - строчки 5-7 с ID пар атомов;
параметры взаимного расположения атомов пары описаны в строчке 9-10.
3 точки могут однозначно расположить сложную структуру в пространстве, поэтому мы выбираем водородные связи как источник данных точек. - что ковалентные связи в гетероатомах нужно вычислять по расстояниям между атомами, строчка 12;
- имя файла с выравниванием и имена последовательностей образца и моделируемого белка, строчка 13 (а имя файла со структурой содержится в выравнивании);
- число и номера моделей, которые нужно построить (в данном примере 5 моделей), строки 14-15;
- что пора строить модель строчка 16.
-
что нужно использовать стандартные валентные углы в полипептидной цепи (строчка 4);
-
Запустим исполнение скрипта:
mod9v7 lysc_human.py &
-
В результате были получены структуры 5 моделей. Они были сохранены в фавйлах model1.pdb, model2.pdb, model3.pdb, model4.pdb и model5.pdb.
Изображение со структурами моделей, наложенными друг на друга, представлено ниже:
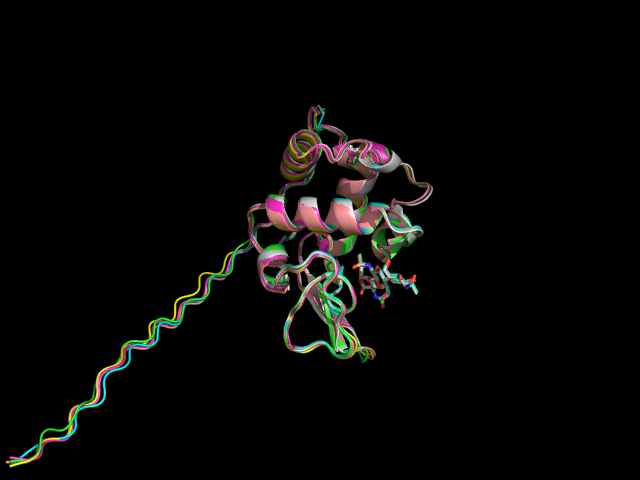
Как видно, структуры моделей практически не отличаются друг от друга (малейшие отличия наблюдаются лишь в петлях). N-конец всех моделей представляет собой длинный хвост. Возник он потому, что в структуре белка-образца не было соответствующих аминокислотных остатков.
-
Проверим качество моделей. Для этого будем использовать сервис WHATIF
Оценим качество карты Рамачандрана (Ramachandran plot evaluation):
Для 1 модели z-score равен 0.264, для 2 - 0.439, для 3 - 0.222, для 4 - 0.021, для 5 - 0.265.
По этому показателю лучшая модель - модель №4.
Оценим длины связей моделей (anomalous bond lengths):
Для 1 модели z-score равен 0.951, для 2 - 0.948, для 3 - 0.955, для 4 - 0.948, для 5 - 0.947.
По этому показателю лучшая модель - модель №5. Впрочем, z-score для всех моделей примерно одинаковы.
Оценим валентные угла моделей (anomalous bond angles):
Для 1 модели z-score равен 1.318 (29 углов с z-score больше 4), для 2 - 1.274 (26 углов с z-score больше 4), для 3 - 1.275 (26 углов с z-score больше 4), для 4 - 1.291 (29 углов с z-score больше 4), для 5 - 1.209 (18 углов с z-score больше 4).
По этому показателю лучшая модель - модель №5.
Все модели очень похожи друг на друга и могут использоваться для дальнейшего анализа белка LYSC_HUMAN, все же лучшей будем считать модель №5.