| Команда | Функция | Результат |
|---|---|---|
| fastqc chr4v0.fastq | Анализ качества необработанного секвенирования | Отчет и zip-архив с его составляющими |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr4v0.fastq chr4trimmed.fastq TRAILING:20 MINLEN:50 | Обрезка с конца чтений нуклеотидов с качеством меньше 20 и последующее отбрасывание чтений короче 50 | Файл chr4trimmed.fastq |
| fastqc chr4trimmed.fastq | Анализ качества обрезанного секвенирования | Отчет и архив с соответсвующими изображениями |
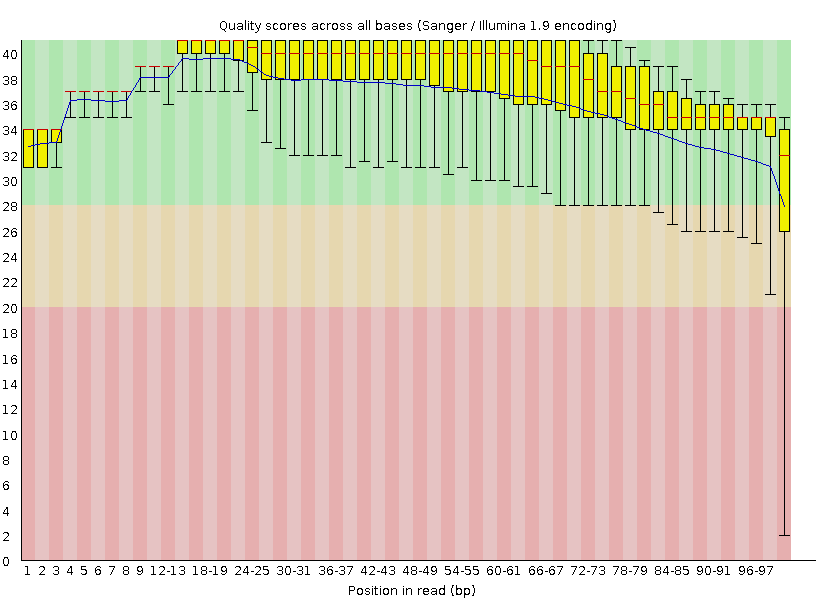
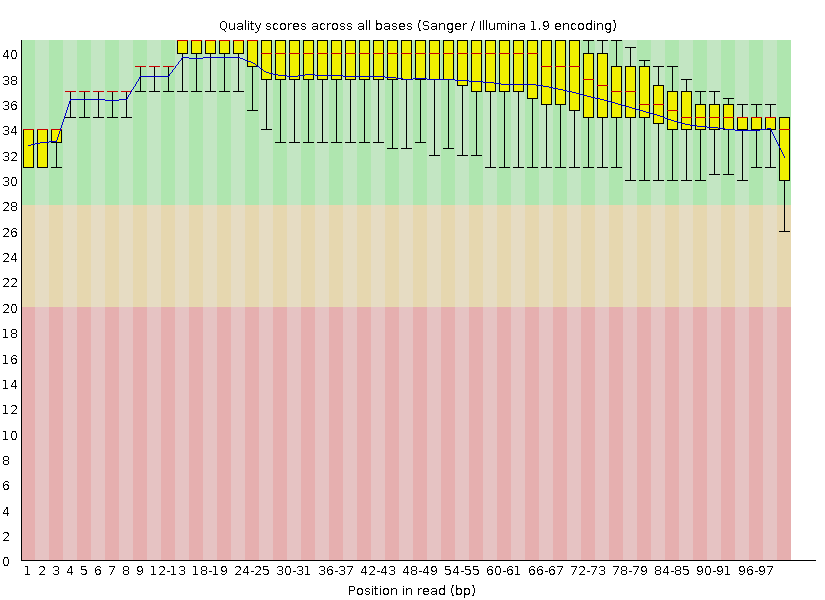
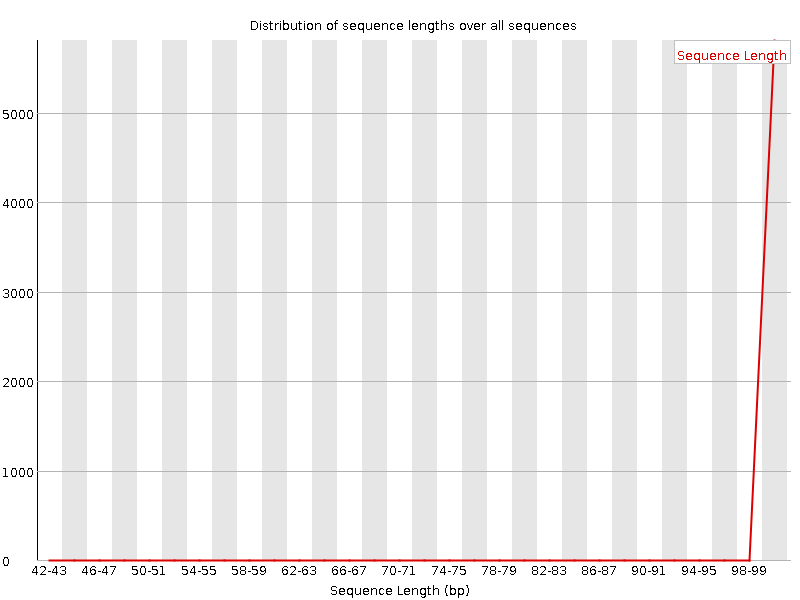
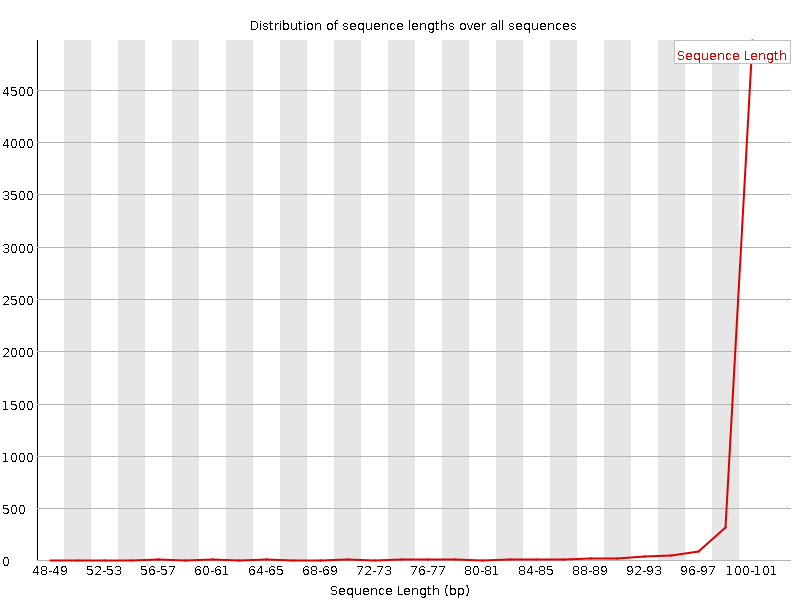
На подготовительном этапе был произведен сбор статистики по начальному материалу, очистка концев и отсев коротких чтений, а потом сбор новой статистики.
| Параметр | Исходное | Подготовленное |
|---|---|---|
| Качество | | |
| Число чтений | 5810 | 5715 |
| Длина чтений | | |
Комментарий: предложенная заданием обработка улучшает картину только в последней четверти длины чтений, сильно уменьшая разброс и среднее. При взгляде на график длин чтений, становится видно отсутствие чтений короче 50 нуклеотидов. Ещё график стал более пологим, из-за того, что прочтения длиной 100 потеряли нуклеотиды на конце и стали более короткими.
| Команда | Функция | Результат |
|---|---|---|
| PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 | Добавление пакета программ в path | Программы становятся дотупны для вызова через командную строку. |
| hisat2-build chr4ref.fasta chr4 | Индексирование референсной последовательности | Несколько файлов расширения ht2 |
| hisat2 -x chr4 -U chr4trimmed.fastq --no-spliced-alignment --no-softclip > 1_align.sam | Создание выравнивания референсной последовательности и прочтений | Выравнивание в формате sam |
| samtools view 1_align.sam -bo 1_align.bam | Перевод выравнивания в бинарный формат | Выравнивание в формате bam |
| samtools sort 1_align.bam -T file.txt -o alignsort.bam | Сортировка бинарного выравнивания | Отсортированное выравнивание |
| samtools index alignsort.bam | Индексирование бинарного выравнивания | Проиндексированное отсортированное выравнивание |
В результате выравнивания 5694 прочтения были мапированы на референс однократно, две более одного раза, а 19 не попали вообще. В sam файле так же была представлена полезная информация о чтениях: ID, координаты на хромосоме, номер хромосомы, количество картрирований.
| Команда | Функция | Результат |
|---|---|---|
| samtools mpileup -uf chr4ref.fasta alignsort.bam -o snp.bcf | Сбор информации о полиморфизмах | Файл с полиморфизмами в бинарном виде |
| bcftools call -cv snp.bcf -o snp.vcf | Перевод последовательности в читаемый вид | Файл с информацией о полиморфизмах |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 /nfs/srv/databases/ngs/sergebus/snp.vcf > /nfs/srv/databases/ngs/sergebus/snp.avinput | Подготовка файла с полиморфизмами к аннотации (индели предварительно удалены вручную) | Подходящий для аннотации исходный файл. |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.snp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация SNP по базе dbsnp | Файл с параметрами программы, всеми и приходящимися на экзоны полиморфизмами. |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.1000g -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация SNP по базе 1000genome | Аналогичные три файла |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация SNP по базе GWAS | Аналогичные три файла |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.clinvar -build hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация SNP по базе clinwar | Аналогичные три файла |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -out rs.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/ | Аннотация SNP по базе refgene | Аналогичные три файла |
Было обнаружено 45 однонуклеотидных полиморфизмов и 4 инделя. Три избранных вариации охарактеризованы в таблице. Конкретно они были выбраны чтобы представить три отличающихся варианта изменений одной поледовательности относительно другой.
| Координата | Референс | Чтение | Природа разногласия | Глубина покрытия | Качество чтений |
|---|---|---|---|---|---|
| 68468036 | c | cAT | Инзерция | 1 | 22.4955 |
| 88759690 | T | C | Замена | 48 | 225.009 |
| 187165891 | gttttt | gtttt | Делеция | 2 | 3.66479 |
Стоит отметить, что аннотация по базе GWAS не дала никаких результатов, а единственный аннотированый по базе clinvar полиморфизм имеет негативный эффект на свертываемость крови (контактный путь). Так как эта мутация, несмотря на нахождение в интроне гена KLKB1, имеет клинические проявления, резонно предположить, что она затрагивает важные для сплайсинга сайты и мутантный белок отличается по экзонному составу. Большая часть SNP попала в интронные участки трех генов. Rs имеют 41 SNP. Максимальная популяционная частота обнаруженных полиморфизмов 91%(замена в 3' нетранслируемом регионе KLKB1), минимальная - 0.3%(замена в интроне гена STAP1). Средняя частота полиморфизмов 39.7%. Не обнаруженные в базах данных полиморфизмы имеют низкое качество. Ниже вы можете ознакомится со всеми SNP.