| Команда | Что делает |
| fastqc chr2.1.fastq | Анализ качества чтений до триммирования |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr2.1.fastq chr2.1_trimmed.fastq TRAILING:20 MINLEN:50 | Триммирование; обрезает с конца чтений нуклеотиды с качеством прочтения меньше 20, удаляет чтения длиной меньше 50 |
| fastqc chr2.1_trimmed.fastq | Анализ качества чтений после триммирования |
Визуализация качетсва ридов. Первое изображение до триммирования, второе - после.
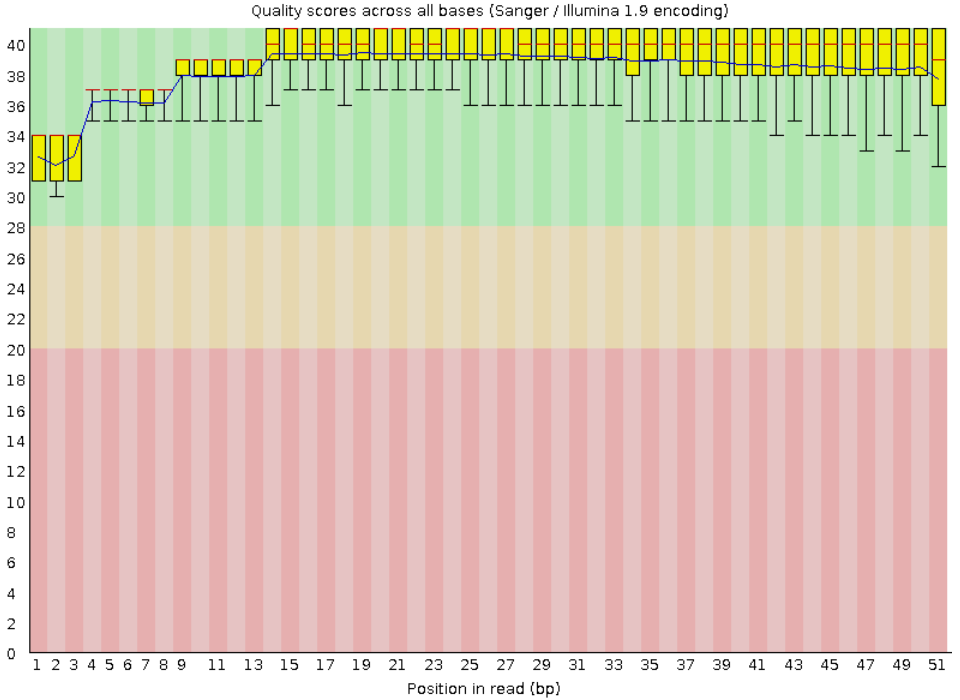
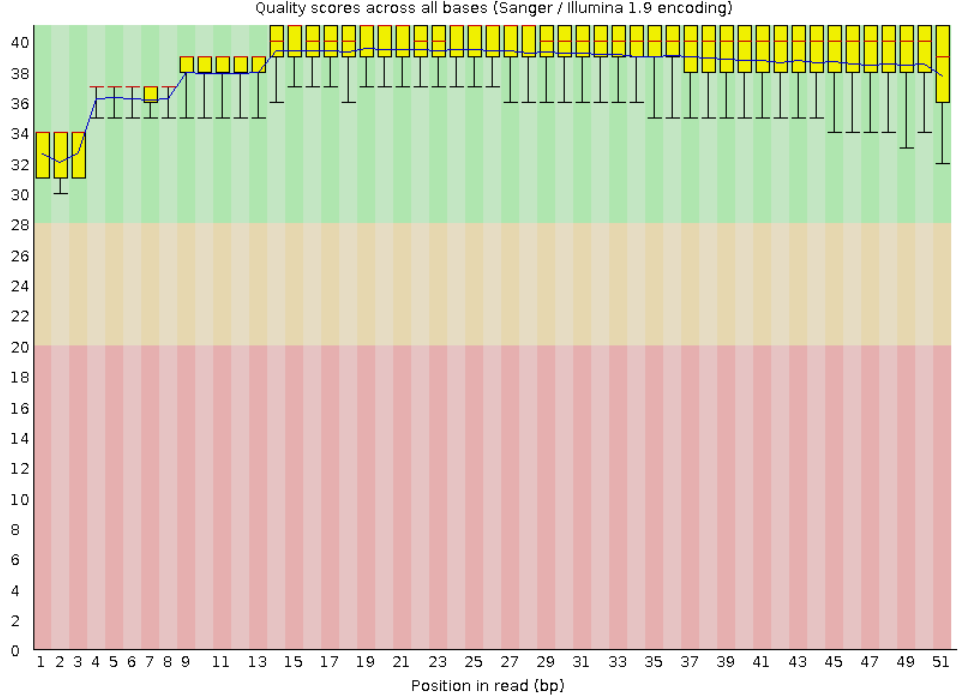
Количество ридов до тримморония: 12 507
Количество ридов после триммирования: 12 399
Вырезалось незначительное количество ридов, из графического представления ясно, что в данном случае триммирование
не было необходимым.
| Команда | Что делает |
| hisat2 -x chr2_ref -U chr2.1_trimmed.fastq -S chr2_alignment.sam --no-softclip 2> hisat_2_2_2.log | Построение выравнивания референсной последовательности и прочтения в формате sam.
-x - индексированная референсная последовательность (была индексирована в предыдущем практикуме) -U - файл с прочтениями -S - название выходного файла Убрала --no-spliced-alignment, потому что мы работаем с последовательностью РНК-траскриптов |
| samtools view -b chr2_alignment.sam -o chr2_alignment.bam | Перевод в бинарный файл
-b - перевод в бинарный формат -о - выходной файл |
| samtools sort chr2_alignment.bam chr2_sorted | Сортировка с референсом |
| samtools index chr2_sorted.bam | Индексирование |
Вывод программы hisat2:
12399 reads; of these:
12399 (100.00%) were unpaired; of these:
54 (0.44%) aligned 0 times
12345 (99.56%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.56% overall alignment rate
Можем заметить, что что 99,56% последовательностей выравнивались с референсом ровно один раз. Качество картирования высокое.
| Команда | Что делает |
| htseq-count -f bam -s no -i gene_id -m union chr2_sorted.bam gencode.v19.chr_patch_hapl_scaff.annotation.gtf > htseqcount_results | Подсчет чтений. -i - задание индекса -s - цепь (yes/reverse/no) -f - формат входного файла (bam/sam) -m - способ подсчета: union; intersection-strict - чтение легло на ген целиком; intersection-nonempty - чтение имеет общую последовательность с геном |
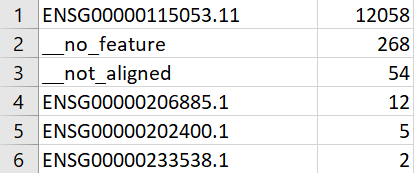
С помощью команды htseq-count был получен файл в формате bam, который был проанализирован с помощью microsoft excel. Использованные
параметры можно посмотреть в таблице выше.
Большинство чтений (12058) легли на ген ENSG00000115053.11, для 268 чтений границы генов не определились, а 54 в принципе не картировались
на хромосому.
Рассмотрим продукты генов, на которые попали риды:
ENSG00000115053.11 - эукариотический ядрышковый фосфопротеин, участвует в сборке и созревании рибосом
ENSG00000206885.1 - малая ядрышковая РНК
ENSG00000202400.1 - малая ядрышковая РНК
ENSG00000233538.1 - видимо, некодирующая РНК