Практикум 2
Филогенетическая реконструкция и сравнение деревьев
Целью данного практикума является реконструировать филогенетическое дерево тремя разными способами и сравнить полученные результаты с филогенетическим деревом, построенном на основе таксономии.
Для начала я создала текстовый файл, состоящий из строк вида: sw:cyb_* , где * - мнемоника одного из организмов, выбранного в предыдущем практикуме.
Также в этот текстовый файл я добавила строку sw:cyb_PANTI. То есть я добавила идентификатор последовательности белка цитохрома B, который принадержит тигру - организму из отряда Хищные.
Зачем я это сделала?
Дело в том, что в филогенетическом дереве, построенном на основе таксономии в ходе предыдущего практикума, имеется неразрешенность: в самом начале дерево разделяется на три ветви (рисунок 1).
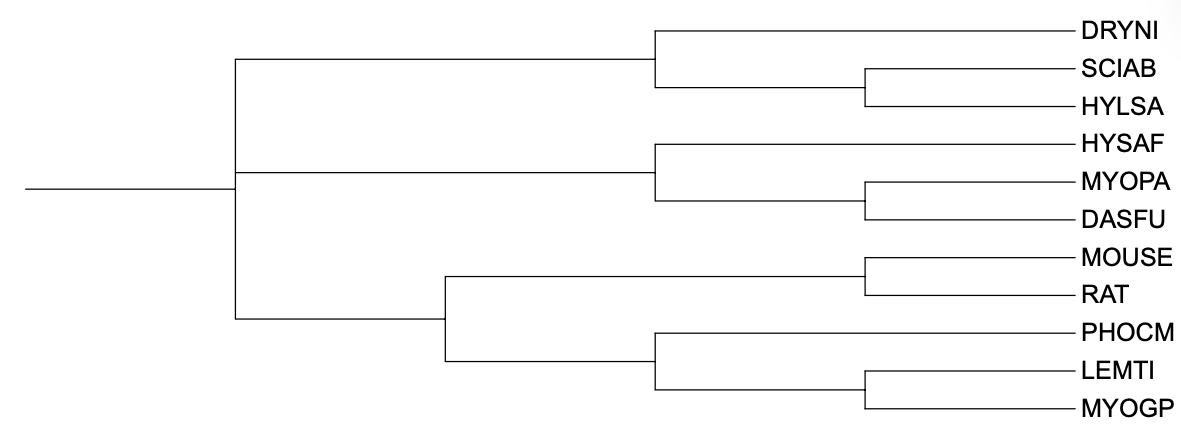 Рисунок 1. Филогенетическое дерево, построенное по таксономии.
Рисунок 1. Филогенетическое дерево, построенное по таксономии.
При визуализации реконструированных деревьев при помощи ITOL необходимо вручную укоренить дерево, то есть в таком случае я самостоятельно должна выбрать, какие две из трех ветвей более сближены между собой. Я посчитала, что это будет неправильно, так как я не имею такой компетенции, поэтому я приняла решение добавить еще один организм, который точно будет более далеким родственником, но при этом несильно. Исходя из этих соображений, я добавила организм из другого отряда Млекопитающих, а именно – из отряда Хищные.
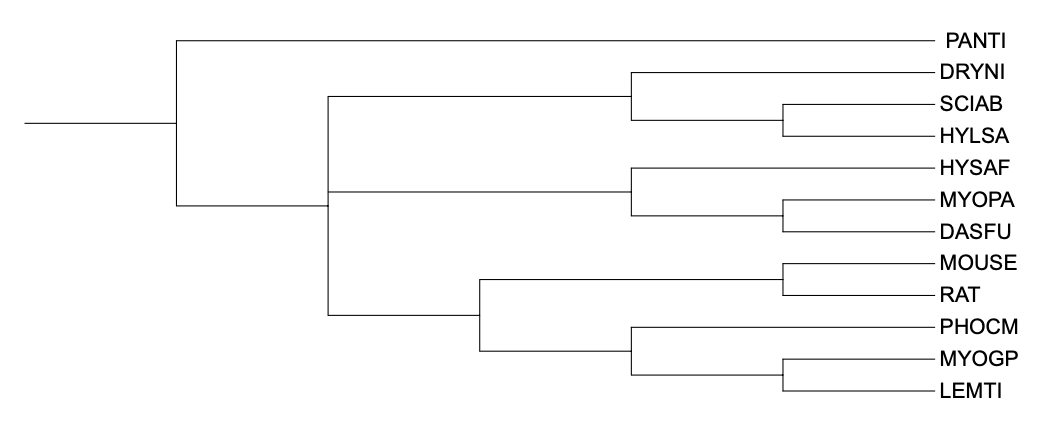 Рисунок 2. Обновленная версия филогенетического дерева, построенного по таксономии.
Рисунок 2. Обновленная версия филогенетического дерева, построенного по таксономии.
После этого я создала файл с последовательностями белка цитохрома B из выбранных организмов в формате fasta при помощи следующей команды:
seqret @cyb.list cyb.fasta
Следующим шагом я выровняла последовательности данных белков при помощи программы muscle:
muscle -align cyb.fasta -output cyb-alignment.fasta
После этого я приступила непосредственно к реконструкции филогенетического дерева.
Реконструкция дерева программой FastME
Сначала я перевела выравнивание в формат phylip-relaxed, читаемый программой FastME, при помощи пакета BioPython.
После этого была проведена непосредственно реконструкция.
p-distance
Для начала в качестве модели была выбрана p-distance:
fastme -pp -i cyb.phy -o tree1p
Далее получившиеся дерево было визуализировано посредством ITOL и укоренено в нужную ветвь. Результат представлен на рисунке 3.
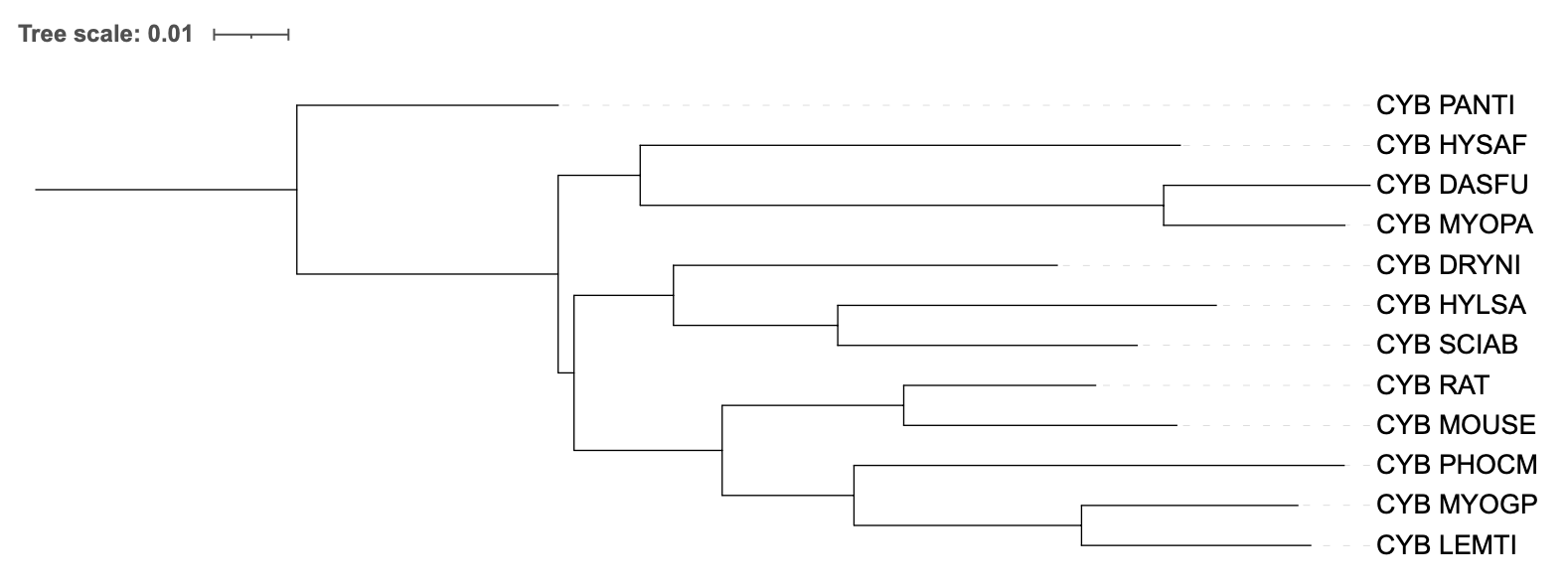 Рисунок 3. Филогенетическое дерево, построенное по таксономии (сверху) и реконструированное программой FastMe, модель – p-distance (снизу).
Рисунок 3. Филогенетическое дерево, построенное по таксономии (сверху) и реконструированное программой FastMe, модель – p-distance (снизу).
Можно заметить, что состав каждой клады в целом остался прежним по сравнению с деревом, построенном по таксономии. Однако интересно, что неразрешенный узел "разрешился"
MtREV
После этого в качестве модели я выбрала MtREV:
fastme -pm -i cyb.phy -o tree1m
Получившиеся дерево было визуализировано посредством ITOL и укоренено в нужную ветвь. Результат представлен на рисунке 4.
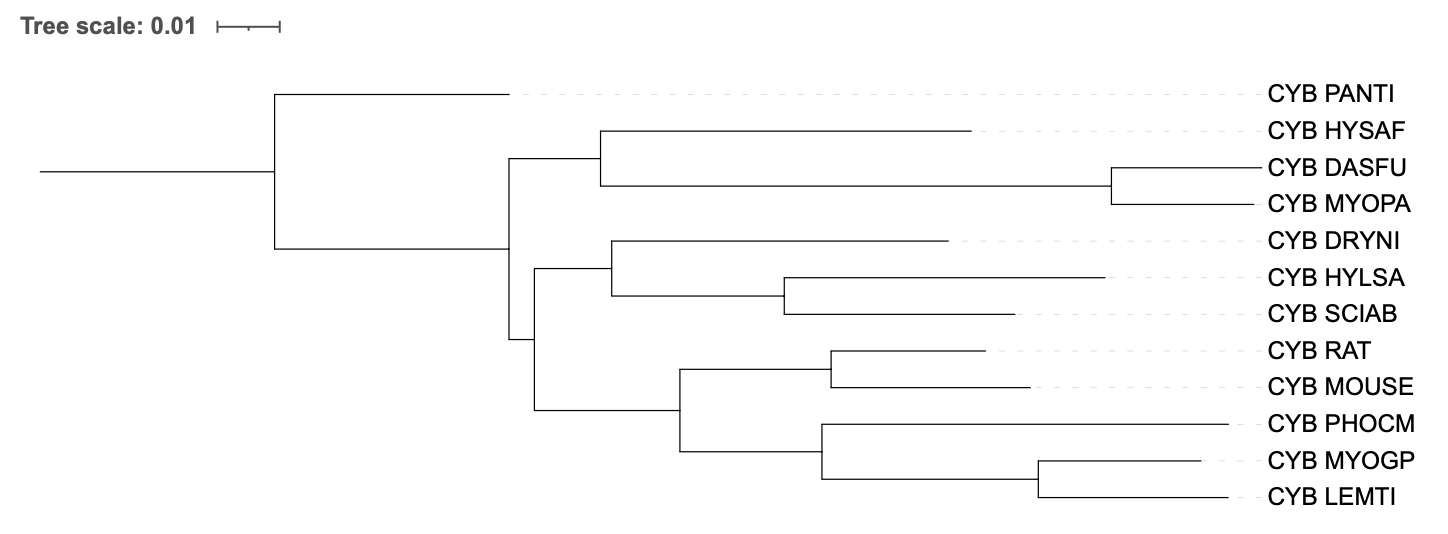 Рисунок 4. Филогенетическое дерево, построенное по таксономии (сверху) и реконструированное программой FastMe, модель – MtREV.
Рисунок 4. Филогенетическое дерево, построенное по таксономии (сверху) и реконструированное программой FastMe, модель – MtREV.
Топология ветвей осталась такой же, как при реконструкции, где в качестве модели была выбрана p-distance. Неразрешенный узел так же "разрешился".
Реконструкция дерева программой IQ-Tree
Программа была запущена с параметрами по умолчанию:
iqtree -s cyb.phy
Полученное дерево было визуализировано при помощи ITOL (рисунок 5).
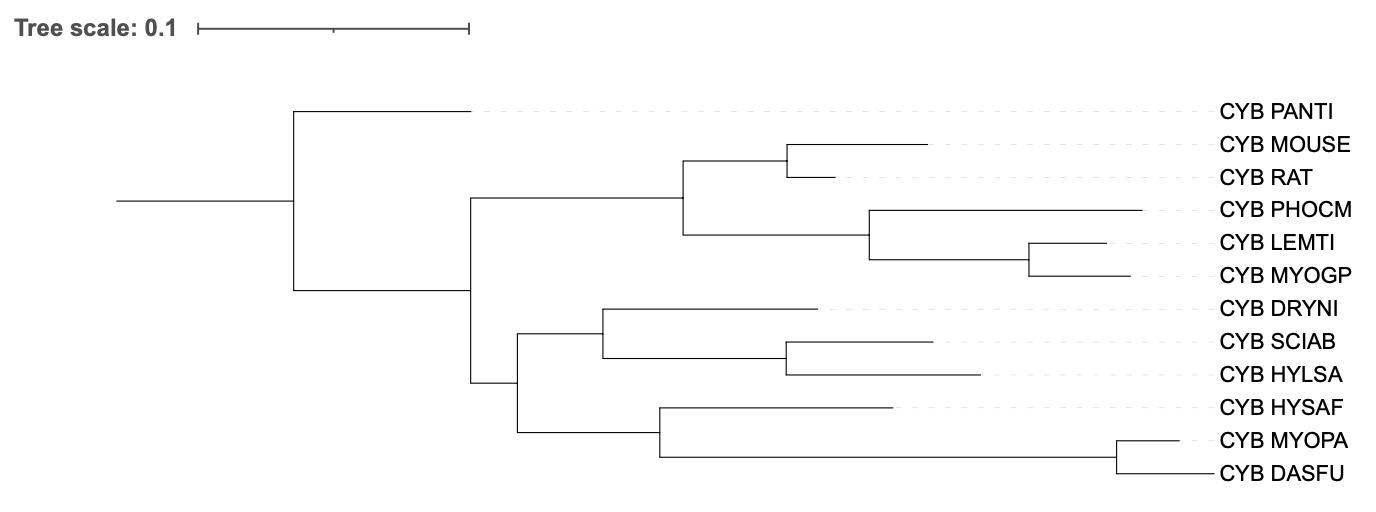 Рисунок 5. Филогенетическое дерево, построенное по таксономии (сверху) и реконструированное программой IQ-Tree (снизу).
Рисунок 5. Филогенетическое дерево, построенное по таксономии (сверху) и реконструированное программой IQ-Tree (снизу).
Можно заметить, что состав трех основных групп и отношение организмов внутри них остались прежними. Однако программа IQ-Tree разрешила узел другим способом.